 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph Neural Network for Metal Organic Framework Potential Energy Approximation
Paper and Code
Oct 29, 2020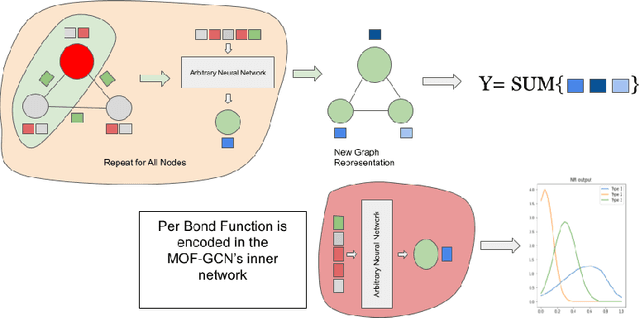
Metal-organic frameworks (MOFs) are nanoporous compounds composed of metal ions and organic linkers. MOFs play an important role in industrial applications such as gas separation, gas purification, and electrolytic catalysis. Important MOF properties such as potential energy are currently computed via techniques such as density functional theory (DFT). Although DFT provides accurate results, it is computationally costly. We propose a machine learning approach for estimating the potential energy of candidate MOFs, decomposing it into separate pair-wise atomic interactions using a graph neural network. Such a technique will allow high-throughput screening of candidates MOFs. We also generate a database of 50,000 spatial configurations and high-quality potential energy values using DFT.