 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTherefore I am. I Think
Apr 02, 2026We consider the question: when a large language reasoning model makes a choice, did it think first and then decide to, or decide first and then think? In this paper, we present evidence that detectable, early-encoded decisions shape chain-of-thought in reasoning models. Specifically, we show that a simple linear probe successfully decodes tool-calling decisions from pre-generation activations with very high confidence, and in some cases, even before a single reasoning token is produced. Activation steering supports this causally: perturbing the decision direction leads to inflated deliberation, and flips behavior in many examples (between 7 - 79% depending on model and benchmark). We also show through behavioral analysis that, when steering changes the decision, the chain-of-thought process often rationalizes the flip rather than resisting it. Together, these results suggest that reasoning models can encode action choices before they begin to deliberate in text.
Distributed Reinforcement Learning for Molecular Design: Antioxidant case
Dec 03, 2023Deep reinforcement learning has successfully been applied for molecular discovery as shown by the Molecule Deep Q-network (MolDQN) algorithm. This algorithm has challenges when applied to optimizing new molecules: training such a model is limited in terms of scalability to larger datasets and the trained model cannot be generalized to different molecules in the same dataset. In this paper, a distributed reinforcement learning algorithm for antioxidants, called DA-MolDQN is proposed to address these problems. State-of-the-art bond dissociation energy (BDE) and ionization potential (IP) predictors are integrated into DA-MolDQN, which are critical chemical properties while optimizing antioxidants. Training time is reduced by algorithmic improvements for molecular modifications. The algorithm is distributed, scalable for up to 512 molecules, and generalizes the model to a diverse set of molecules. The proposed models are trained with a proprietary antioxidant dataset. The results have been reproduced with both proprietary and public datasets. The proposed molecules have been validated with DFT simulations and a subset of them confirmed in public "unseen" datasets. In summary, DA-MolDQN is up to 100x faster than previous algorithms and can discover new optimized molecules from proprietary and public antioxidants.
STRIDE: Structure-guided Generation for Inverse Design of Molecules
Nov 06, 2023Machine learning and especially deep learning has had an increasing impact on molecule and materials design. In particular, given the growing access to an abundance of high-quality small molecule data for generative modeling for drug design, results for drug discovery have been promising. However, for many important classes of materials such as catalysts, antioxidants, and metal-organic frameworks, such large datasets are not available. Such families of molecules with limited samples and structural similarities are especially prevalent for industrial applications. As is well-known, retraining and even fine-tuning are challenging on such small datasets. Novel, practically applicable molecules are most often derivatives of well-known molecules, suggesting approaches to addressing data scarcity. To address this problem, we introduce $\textbf{STRIDE}$, a generative molecule workflow that generates novel molecules with an unconditional generative model guided by known molecules without any retraining. We generate molecules outside of the training data from a highly specialized set of antioxidant molecules. Our generated molecules have on average 21.7% lower synthetic accessibility scores and also reduce ionization potential by 5.9% of generated molecules via guiding.
ParticleGrid: Enabling Deep Learning using 3D Representation of Materials
Nov 15, 2022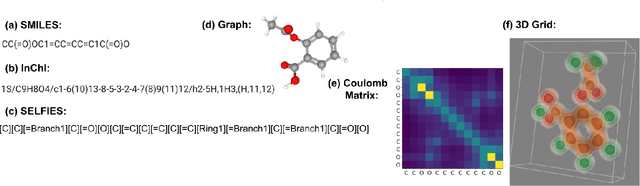
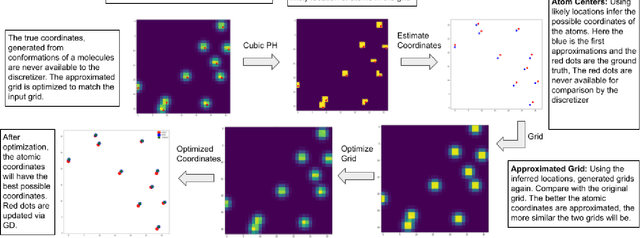
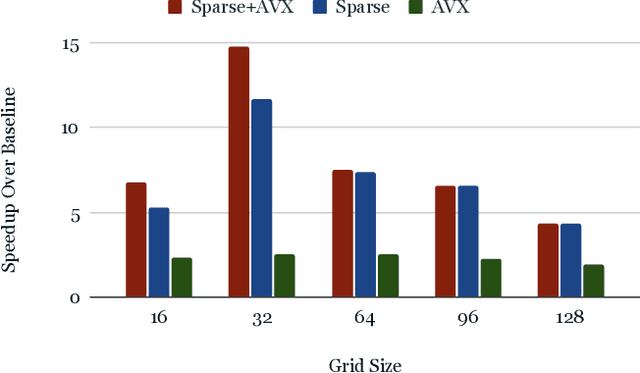
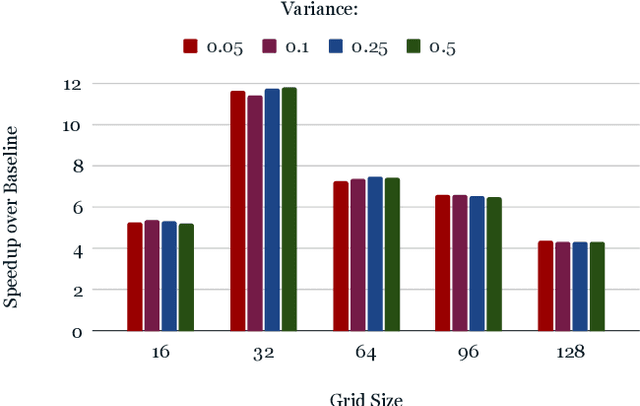
From AlexNet to Inception, autoencoders to diffusion models, the development of novel and powerful deep learning models and learning algorithms has proceeded at breakneck speeds. In part, we believe that rapid iteration of model architecture and learning techniques by a large community of researchers over a common representation of the underlying entities has resulted in transferable deep learning knowledge. As a result, model scale, accuracy, fidelity, and compute performance have dramatically increased in computer vision and natural language processing. On the other hand, the lack of a common representation for chemical structure has hampered similar progress. To enable transferable deep learning, we identify the need for a robust 3-dimensional representation of materials such as molecules and crystals. The goal is to enable both materials property prediction and materials generation with 3D structures. While computationally costly, such representations can model a large set of chemical structures. We propose $\textit{ParticleGrid}$, a SIMD-optimized library for 3D structures, that is designed for deep learning applications and to seamlessly integrate with deep learning frameworks. Our highly optimized grid generation allows for generating grids on the fly on the CPU, reducing storage and GPU compute and memory requirements. We show the efficacy of 3D grids generated via $\textit{ParticleGrid}$ and accurately predict molecular energy properties using a 3D convolutional neural network. Our model is able to get 0.006 mean square error and nearly match the values calculated using computationally costly density functional theory at a fraction of the time.