 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSystem of Agentic AI for the Discovery of Metal-Organic Frameworks
Apr 18, 2025Generative models and machine learning promise accelerated material discovery in MOFs for CO2 capture and water harvesting but face significant challenges navigating vast chemical spaces while ensuring synthetizability. Here, we present MOFGen, a system of Agentic AI comprising interconnected agents: a large language model that proposes novel MOF compositions, a diffusion model that generates crystal structures, quantum mechanical agents that optimize and filter candidates, and synthetic-feasibility agents guided by expert rules and machine learning. Trained on all experimentally reported MOFs and computational databases, MOFGen generated hundreds of thousands of novel MOF structures and synthesizable organic linkers. Our methodology was validated through high-throughput experiments and the successful synthesis of five "AI-dreamt" MOFs, representing a major step toward automated synthesizable material discovery.
Cross-functional transferability in universal machine learning interatomic potentials
Apr 07, 2025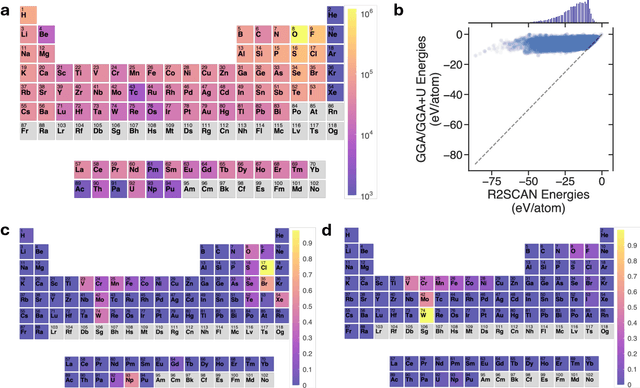



The rapid development of universal machine learning interatomic potentials (uMLIPs) has demonstrated the possibility for generalizable learning of the universal potential energy surface. In principle, the accuracy of uMLIPs can be further improved by bridging the model from lower-fidelity datasets to high-fidelity ones. In this work, we analyze the challenge of this transfer learning problem within the CHGNet framework. We show that significant energy scale shifts and poor correlations between GGA and r$^2$SCAN pose challenges to cross-functional data transferability in uMLIPs. By benchmarking different transfer learning approaches on the MP-r$^2$SCAN dataset of 0.24 million structures, we demonstrate the importance of elemental energy referencing in the transfer learning of uMLIPs. By comparing the scaling law with and without the pre-training on a low-fidelity dataset, we show that significant data efficiency can still be achieved through transfer learning, even with a target dataset of sub-million structures. We highlight the importance of proper transfer learning and multi-fidelity learning in creating next-generation uMLIPs on high-fidelity data.
Large Language Models Are Innate Crystal Structure Generators
Feb 28, 2025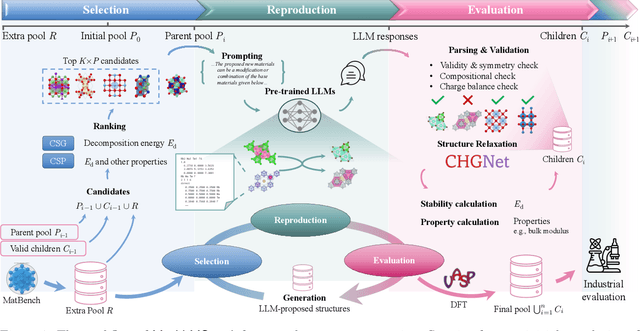
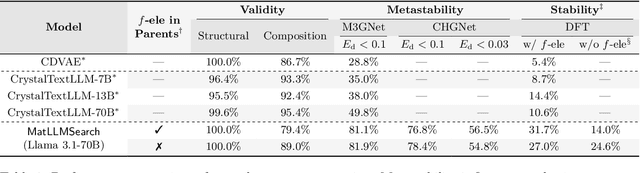
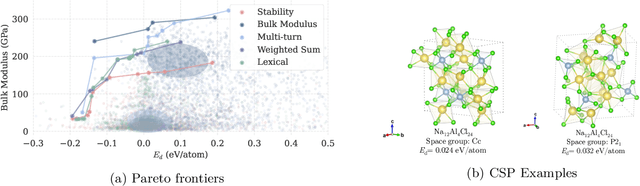
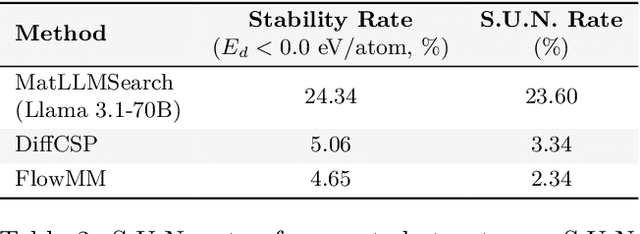
Crystal structure generation is fundamental to materials discovery, enabling the prediction of novel materials with desired properties. While existing approaches leverage Large Language Models (LLMs) through extensive fine-tuning on materials databases, we show that pre-trained LLMs can inherently generate stable crystal structures without additional training. Our novel framework MatLLMSearch integrates pre-trained LLMs with evolutionary search algorithms, achieving a 78.38% metastable rate validated by machine learning interatomic potentials and 31.7% DFT-verified stability via quantum mechanical calculations, outperforming specialized models such as CrystalTextLLM. Beyond crystal structure generation, we further demonstrate that our framework can be readily adapted to diverse materials design tasks, including crystal structure prediction and multi-objective optimization of properties such as deformation energy and bulk modulus, all without fine-tuning. These results establish pre-trained LLMs as versatile and effective tools for materials discovery, opening up new venues for crystal structure generation with reduced computational overhead and broader accessibility.
Overcoming systematic softening in universal machine learning interatomic potentials by fine-tuning
May 11, 2024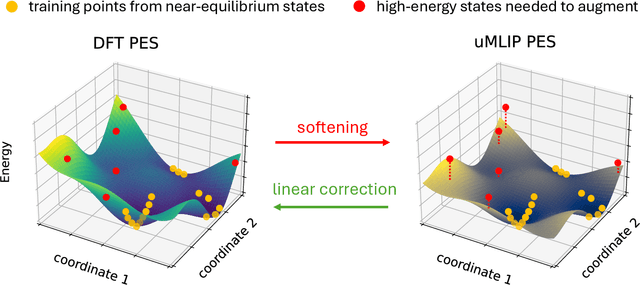
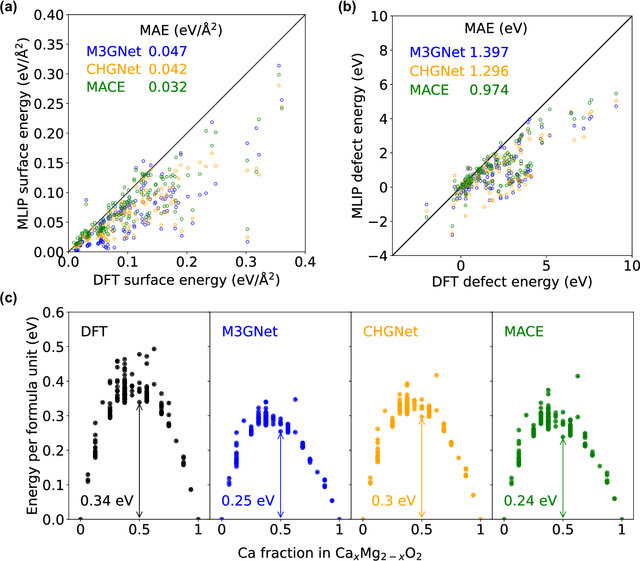
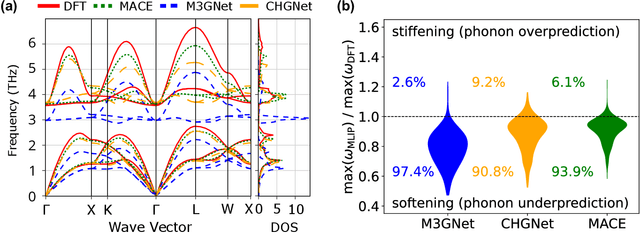
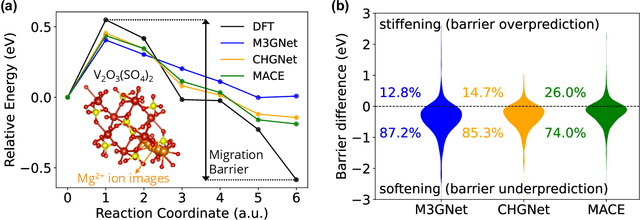
Machine learning interatomic potentials (MLIPs) have introduced a new paradigm for atomic simulations. Recent advancements have seen the emergence of universal MLIPs (uMLIPs) that are pre-trained on diverse materials datasets, providing opportunities for both ready-to-use universal force fields and robust foundations for downstream machine learning refinements. However, their performance in extrapolating to out-of-distribution complex atomic environments remains unclear. In this study, we highlight a consistent potential energy surface (PES) softening effect in three uMLIPs: M3GNet, CHGNet, and MACE-MP-0, which is characterized by energy and force under-prediction in a series of atomic-modeling benchmarks including surfaces, defects, solid-solution energetics, phonon vibration modes, ion migration barriers, and general high-energy states. We find that the PES softening behavior originates from a systematic underprediction error of the PES curvature, which derives from the biased sampling of near-equilibrium atomic arrangements in uMLIP pre-training datasets. We demonstrate that the PES softening issue can be effectively rectified by fine-tuning with a single additional data point. Our findings suggest that a considerable fraction of uMLIP errors are highly systematic, and can therefore be efficiently corrected. This result rationalizes the data-efficient fine-tuning performance boost commonly observed with foundational MLIPs. We argue for the importance of a comprehensive materials dataset with improved PES sampling for next-generation foundational MLIPs.
Matbench Discovery -- An evaluation framework for machine learning crystal stability prediction
Aug 28, 2023Matbench Discovery simulates the deployment of machine learning (ML) energy models in a high-throughput search for stable inorganic crystals. We address the disconnect between (i) thermodynamic stability and formation energy and (ii) in-domain vs out-of-distribution performance. Alongside this paper, we publish a Python package to aid with future model submissions and a growing online leaderboard with further insights into trade-offs between various performance metrics. To answer the question which ML methodology performs best at materials discovery, our initial release explores a variety of models including random forests, graph neural networks (GNN), one-shot predictors, iterative Bayesian optimizers and universal interatomic potentials (UIP). Ranked best-to-worst by their test set F1 score on thermodynamic stability prediction, we find CHGNet > M3GNet > MACE > ALIGNN > MEGNet > CGCNN > CGCNN+P > Wrenformer > BOWSR > Voronoi tessellation fingerprints with random forest. The top 3 models are UIPs, the winning methodology for ML-guided materials discovery, achieving F1 scores of ~0.6 for crystal stability classification and discovery acceleration factors (DAF) of up to 5x on the first 10k most stable predictions compared to dummy selection from our test set. We also highlight a sharp disconnect between commonly used global regression metrics and more task-relevant classification metrics. Accurate regressors are susceptible to unexpectedly high false-positive rates if those accurate predictions lie close to the decision boundary at 0 eV/atom above the convex hull where most materials are. Our results highlight the need to focus on classification metrics that actually correlate with improved stability hit rate.
Extracting Structured Seed-Mediated Gold Nanorod Growth Procedures from Literature with GPT-3
Apr 26, 2023Although gold nanorods have been the subject of much research, the pathways for controlling their shape and thereby their optical properties remain largely heuristically understood. Although it is apparent that the simultaneous presence of and interaction between various reagents during synthesis control these properties, computational and experimental approaches for exploring the synthesis space can be either intractable or too time-consuming in practice. This motivates an alternative approach leveraging the wealth of synthesis information already embedded in the body of scientific literature by developing tools to extract relevant structured data in an automated, high-throughput manner. To that end, we present an approach using the powerful GPT-3 language model to extract structured multi-step seed-mediated growth procedures and outcomes for gold nanorods from unstructured scientific text. GPT-3 prompt completions are fine-tuned to predict synthesis templates in the form of JSON documents from unstructured text input with an overall accuracy of $86\%$. The performance is notable, considering the model is performing simultaneous entity recognition and relation extraction. We present a dataset of 11,644 entities extracted from 1,137 papers, resulting in 268 papers with at least one complete seed-mediated gold nanorod growth procedure and outcome for a total of 332 complete procedures.