 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAgentic LLM Reasoning in a Self-Driving Laboratory for Air-Sensitive Lithium Halide Spinel Conductors
Apr 13, 2026Self-driving laboratories promise to accelerate materials discovery. Yet current automated solid-state synthesis platforms are limited to ambient conditions, thereby precluding their use for air-sensitive materials. Here, we present A-Lab for Glovebox Powder Solid-state Synthesis (A-Lab GPSS), a robotic platform capable of synthesizing and characterizing air-sensitive inorganic materials under strict air-free conditions. By integrating an agentic AI framework into the A-Lab GPSS platform, we structure autonomous experimental design through abductive and inductive reasoning. We deploy this platform to explore the vast compositional space of lithium halide spinel solid-state ionic conductors. Across a synthesis campaign comprising 352 samples with diverse compositions, the system explores a broad chemical space, experimentally realizing 72% of the 171 possible pairwise combinations among the 19 metals considered in this study. Over the course of the campaign, the fraction of compositions exhibiting both good ionic conductivity (> 0.05 mS/cm) and high halide spinel phase purity increases from 1.33% in the first 75 agent-proposed samples to 5.33% in the final 75. Furthermore, by inspecting the AI's reasoning processes, we reveal distinct yet complementary discovery strategies: abductive reasoning interrogates abnormal observations within already explored regions, whereas inductive reasoning expands the search into broader, previously unvisited chemical space. This work establishes a scalable platform for the autonomous discovery of complex, air-sensitive solid-state materials.
MATRIX: A Multimodal Benchmark and Post-Training Framework for Materials Science
Jan 30, 2026Scientific reasoning in materials science requires integrating multimodal experimental evidence with underlying physical theory. Existing benchmarks make it difficult to assess whether incorporating visual experimental data during post-training improves mechanism-grounded explanation reasoning beyond text-only supervision. We introduce MATRIX, a multimodal benchmark for materials science reasoning that evaluates foundational theory, research-level reasoning, and the interpretation of real experimental artifacts across multiple characterization modalities. Using MATRIX as a controlled diagnostic, we isolate the effect of visual grounding by comparing post-training on structured materials science text alone with post-training that incorporates paired experimental images. Despite using relatively small amounts of multimodal data, visual supervision improves experimental interpretation by 10-25% and yields 5-16% gains on text-only scientific reasoning tasks. Our results demonstrate that these improvements rely on correct image-text alignment during post-training, highlighting cross-modal representational transfer. We also observe consistent improvements on ScienceQA and PubMedQA, demonstrating that the benefits of structured multimodal post-training extend beyond materials science. The MATRIX dataset is available at https://huggingface.co/datasets/radical-ai/MATRIX and the model at https://huggingface.co/radical-ai/MATRIX-PT.
CASCADE: Cumulative Agentic Skill Creation through Autonomous Development and Evolution
Dec 29, 2025Large language model (LLM) agents currently depend on predefined tools or brittle tool generation, constraining their capability and adaptability to complex scientific tasks. We introduce CASCADE, a self-evolving agentic framework representing an early instantiation of the transition from "LLM + tool use" to "LLM + skill acquisition". CASCADE enables agents to master complex external tools and codify knowledge through two meta-skills: continuous learning via web search and code extraction, and self-reflection via introspection and knowledge graph exploration, among others. We evaluate CASCADE on SciSkillBench, a benchmark of 116 materials science and chemistry research tasks. CASCADE achieves a 93.3% success rate using GPT-5, compared to 35.4% without evolution mechanisms. We further demonstrate real-world applications in computational analysis, autonomous laboratory experiments, and selective reproduction of published papers. Along with human-agent collaboration and memory consolidation, CASCADE accumulates executable skills that can be shared across agents and scientists, moving toward scalable AI-assisted scientific research.
Cross-functional transferability in universal machine learning interatomic potentials
Apr 07, 2025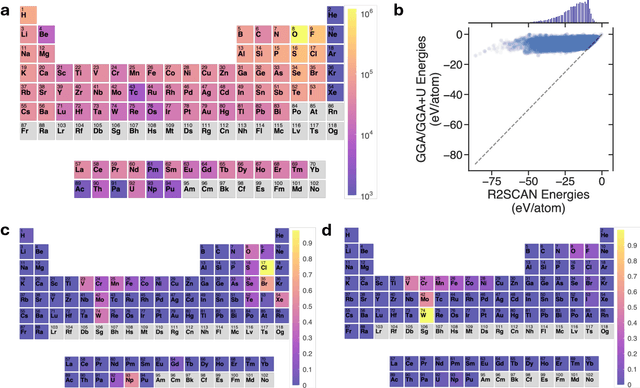



The rapid development of universal machine learning interatomic potentials (uMLIPs) has demonstrated the possibility for generalizable learning of the universal potential energy surface. In principle, the accuracy of uMLIPs can be further improved by bridging the model from lower-fidelity datasets to high-fidelity ones. In this work, we analyze the challenge of this transfer learning problem within the CHGNet framework. We show that significant energy scale shifts and poor correlations between GGA and r$^2$SCAN pose challenges to cross-functional data transferability in uMLIPs. By benchmarking different transfer learning approaches on the MP-r$^2$SCAN dataset of 0.24 million structures, we demonstrate the importance of elemental energy referencing in the transfer learning of uMLIPs. By comparing the scaling law with and without the pre-training on a low-fidelity dataset, we show that significant data efficiency can still be achieved through transfer learning, even with a target dataset of sub-million structures. We highlight the importance of proper transfer learning and multi-fidelity learning in creating next-generation uMLIPs on high-fidelity data.
A practical guide to machine learning interatomic potentials -- Status and future
Mar 12, 2025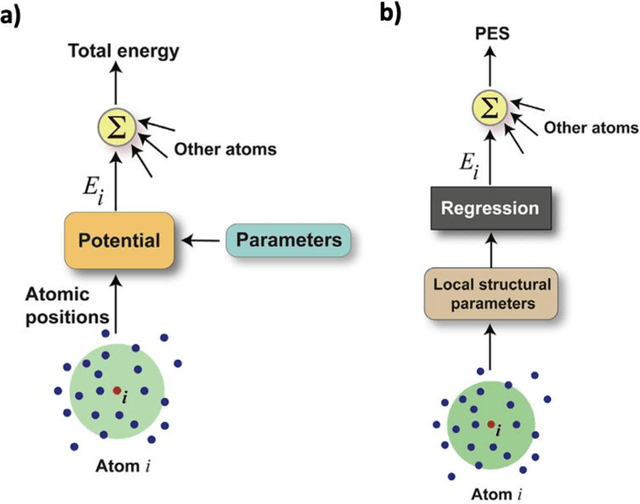

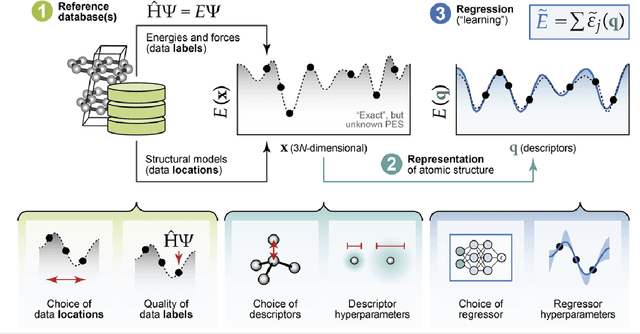
The rapid development and large body of literature on machine learning interatomic potentials (MLIPs) can make it difficult to know how to proceed for researchers who are not experts but wish to use these tools. The spirit of this review is to help such researchers by serving as a practical, accessible guide to the state-of-the-art in MLIPs. This review paper covers a broad range of topics related to MLIPs, including (i) central aspects of how and why MLIPs are enablers of many exciting advancements in molecular modeling, (ii) the main underpinnings of different types of MLIPs, including their basic structure and formalism, (iii) the potentially transformative impact of universal MLIPs for both organic and inorganic systems, including an overview of the most recent advances, capabilities, downsides, and potential applications of this nascent class of MLIPs, (iv) a practical guide for estimating and understanding the execution speed of MLIPs, including guidance for users based on hardware availability, type of MLIP used, and prospective simulation size and time, (v) a manual for what MLIP a user should choose for a given application by considering hardware resources, speed requirements, energy and force accuracy requirements, as well as guidance for choosing pre-trained potentials or fitting a new potential from scratch, (vi) discussion around MLIP infrastructure, including sources of training data, pre-trained potentials, and hardware resources for training, (vii) summary of some key limitations of present MLIPs and current approaches to mitigate such limitations, including methods of including long-range interactions, handling magnetic systems, and treatment of excited states, and finally (viii) we finish with some more speculative thoughts on what the future holds for the development and application of MLIPs over the next 3-10+ years.
AlabOS: A Python-based Reconfigurable Workflow Management Framework for Autonomous Laboratories
May 22, 2024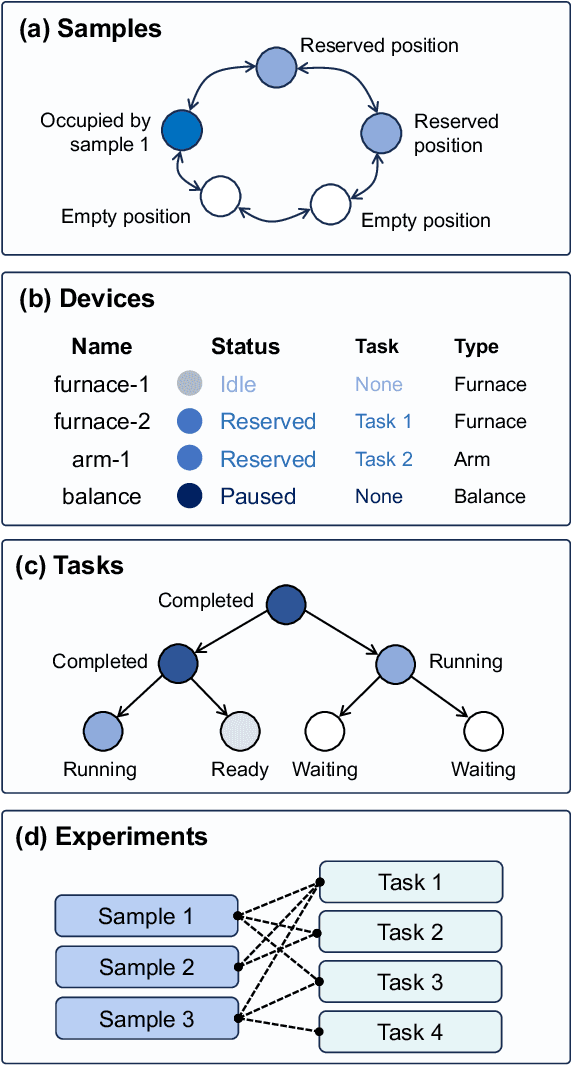
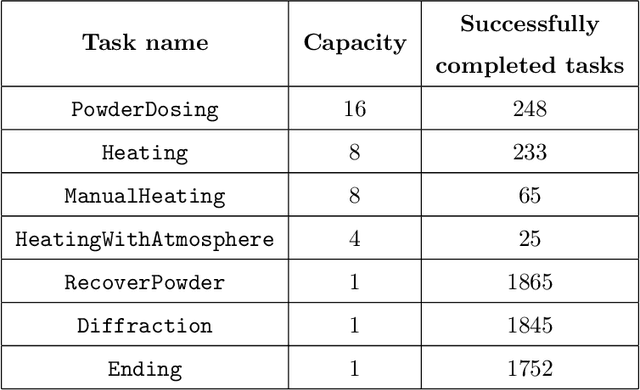
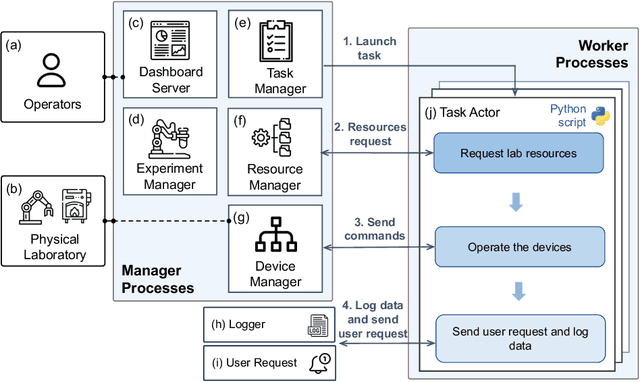
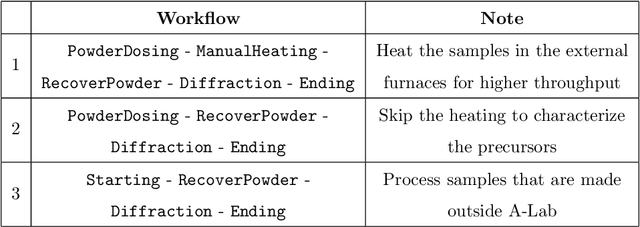
The recent advent of autonomous laboratories, coupled with algorithms for high-throughput screening and active learning, promises to accelerate materials discovery and innovation. As these autonomous systems grow in complexity, the demand for robust and efficient workflow management software becomes increasingly critical. In this paper, we introduce AlabOS, a general-purpose software framework for orchestrating experiments and managing resources, with an emphasis on automated laboratories for materials synthesis and characterization. We demonstrate the implementation of AlabOS in a prototype autonomous materials laboratory. AlabOS features a reconfigurable experiment workflow model, enabling the simultaneous execution of varied workflows composed of modular tasks. Therefore, AlabOS is well-suited to handle the rapidly changing experimental protocols defining the progress of self-driving laboratory development for materials research.
Overcoming systematic softening in universal machine learning interatomic potentials by fine-tuning
May 11, 2024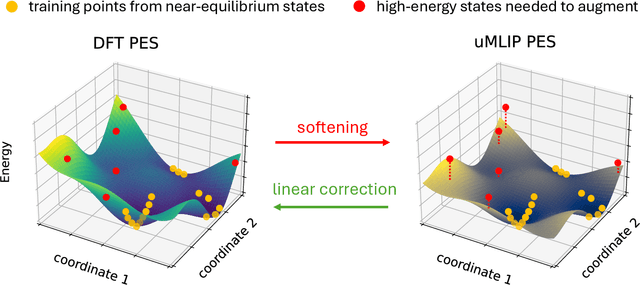
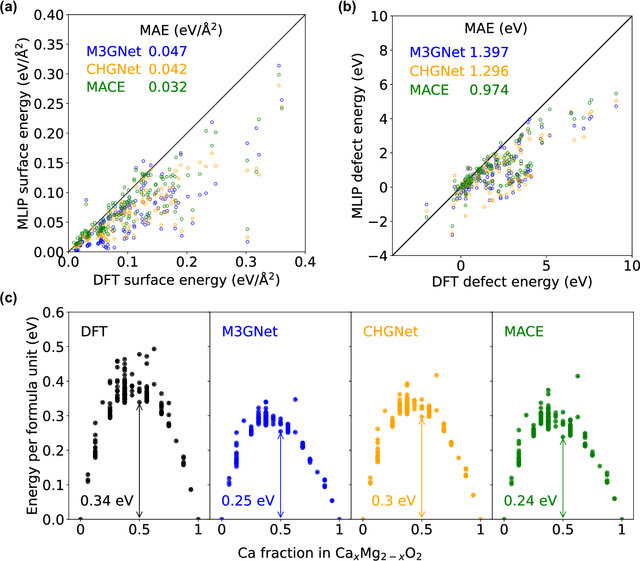
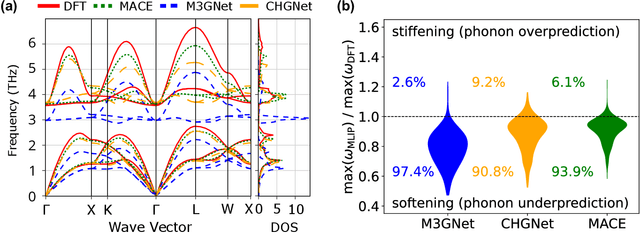
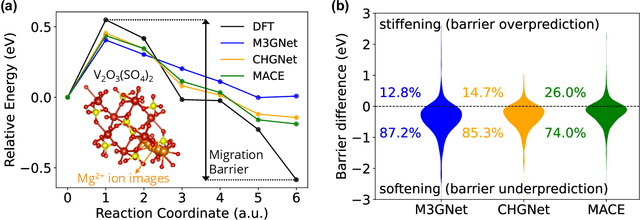
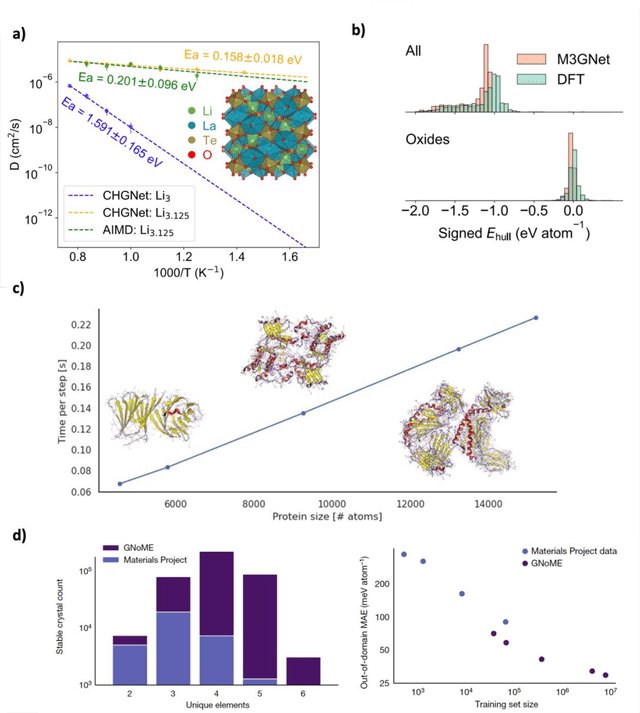
Machine learning interatomic potentials (MLIPs) have introduced a new paradigm for atomic simulations. Recent advancements have seen the emergence of universal MLIPs (uMLIPs) that are pre-trained on diverse materials datasets, providing opportunities for both ready-to-use universal force fields and robust foundations for downstream machine learning refinements. However, their performance in extrapolating to out-of-distribution complex atomic environments remains unclear. In this study, we highlight a consistent potential energy surface (PES) softening effect in three uMLIPs: M3GNet, CHGNet, and MACE-MP-0, which is characterized by energy and force under-prediction in a series of atomic-modeling benchmarks including surfaces, defects, solid-solution energetics, phonon vibration modes, ion migration barriers, and general high-energy states. We find that the PES softening behavior originates from a systematic underprediction error of the PES curvature, which derives from the biased sampling of near-equilibrium atomic arrangements in uMLIP pre-training datasets. We demonstrate that the PES softening issue can be effectively rectified by fine-tuning with a single additional data point. Our findings suggest that a considerable fraction of uMLIP errors are highly systematic, and can therefore be efficiently corrected. This result rationalizes the data-efficient fine-tuning performance boost commonly observed with foundational MLIPs. We argue for the importance of a comprehensive materials dataset with improved PES sampling for next-generation foundational MLIPs.
Extracting Structured Seed-Mediated Gold Nanorod Growth Procedures from Literature with GPT-3
Apr 26, 2023Although gold nanorods have been the subject of much research, the pathways for controlling their shape and thereby their optical properties remain largely heuristically understood. Although it is apparent that the simultaneous presence of and interaction between various reagents during synthesis control these properties, computational and experimental approaches for exploring the synthesis space can be either intractable or too time-consuming in practice. This motivates an alternative approach leveraging the wealth of synthesis information already embedded in the body of scientific literature by developing tools to extract relevant structured data in an automated, high-throughput manner. To that end, we present an approach using the powerful GPT-3 language model to extract structured multi-step seed-mediated growth procedures and outcomes for gold nanorods from unstructured scientific text. GPT-3 prompt completions are fine-tuned to predict synthesis templates in the form of JSON documents from unstructured text input with an overall accuracy of $86\%$. The performance is notable, considering the model is performing simultaneous entity recognition and relation extraction. We present a dataset of 11,644 entities extracted from 1,137 papers, resulting in 268 papers with at least one complete seed-mediated gold nanorod growth procedure and outcome for a total of 332 complete procedures.
CHGNet: Pretrained universal neural network potential for charge-informed atomistic modeling
Feb 28, 2023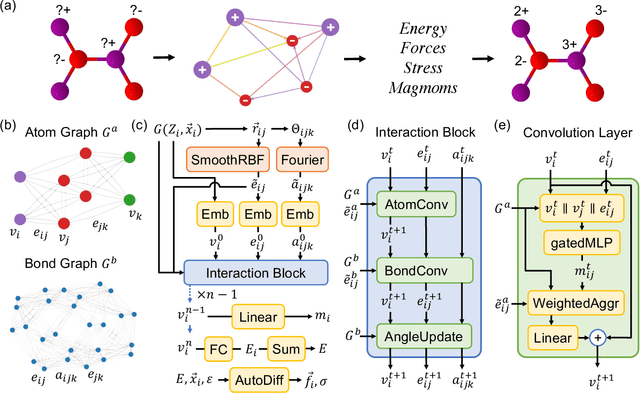
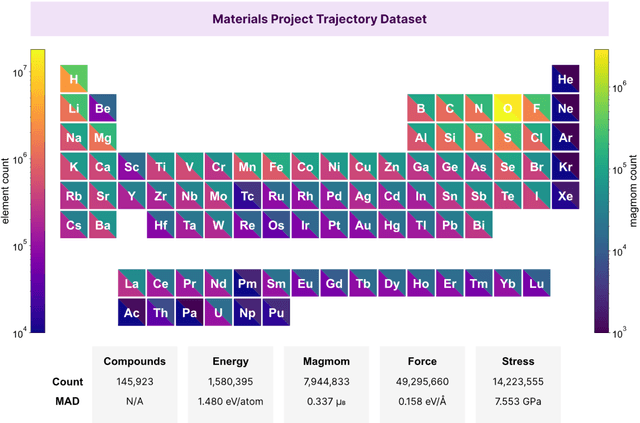
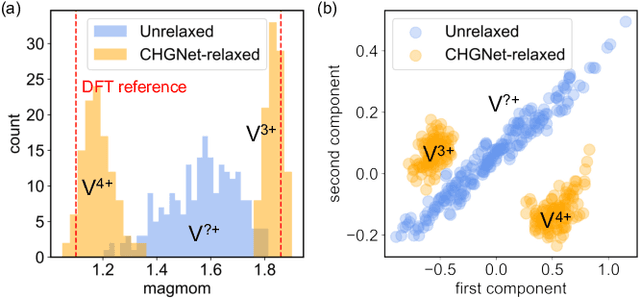
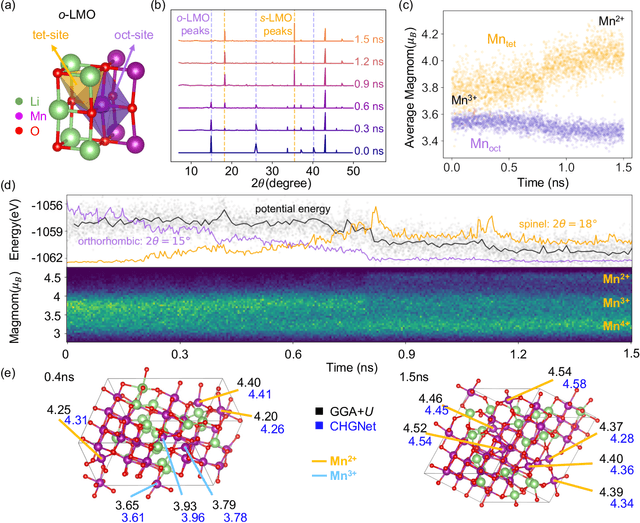
The simulation of large-scale systems with complex electron interactions remains one of the greatest challenges for the atomistic modeling of materials. Although classical force-fields often fail to describe the coupling between electronic states and ionic rearrangements, the more accurate \textit{ab-initio} molecular dynamics suffers from computational complexity that prevents long-time and large-scale simulations, which are essential to study many technologically relevant phenomena, such as reactions, ion migrations, phase transformations, and degradation. In this work, we present the Crystal Hamiltonian Graph neural Network (CHGNet) as a novel machine-learning interatomic potential (MLIP), using a graph-neural-network-based force-field to model a universal potential energy surface. CHGNet is pretrained on the energies, forces, stresses, and magnetic moments from the Materials Project Trajectory Dataset, which consists of over 10 years of density functional theory static and relaxation trajectories of $\sim 1.5$ million inorganic structures. The explicit inclusion of magnetic moments enables CHGNet to learn and accurately represent the orbital occupancy of electrons, enhancing its capability to describe both atomic and electronic degrees of freedom. We demonstrate several applications of CHGNet in solid-state materials, including charge-informed molecular dynamics in Li$_x$MnO$_2$, the finite temperature phase diagram for Li$_x$FePO$_4$ and Li diffusion in garnet conductors. We critically analyze the significance of including charge information for capturing appropriate chemistry, and we provide new insights into ionic systems with additional electronic degrees of freedom that can not be observed by previous MLIPs.
Inorganic synthesis recommendation by machine learning materials similarity from scientific literature
Feb 05, 2023Synthesis prediction is a key accelerator for the rapid design of advanced materials. However, determining synthesis variables such as the choice of precursor materials, operations, and conditions is challenging for inorganic materials because the sequence of reactions during heating is not well understood. In this work, we use a knowledge base of 29,900 solid-state synthesis recipes, text-mined from the scientific literature, to automatically learn which precursors to recommend for the synthesis of a novel target material. The data-driven approach learns chemical similarity of materials and refers the synthesis of a new target to precedent synthesis procedures of similar materials, mimicking human synthesis design. When proposing five precursor sets for each of 2,654 unseen test target materials, the recommendation strategy achieves a success rate of at least 82%. Our approach captures decades of heuristic synthesis data in a mathematical form, making it accessible for use in recommendation engines and autonomous laboratories.