 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInvestigating the Behavior of Diffusion Models for Accelerating Electronic Structure Calculations
Nov 02, 2023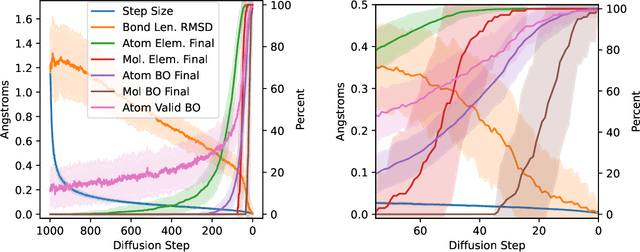
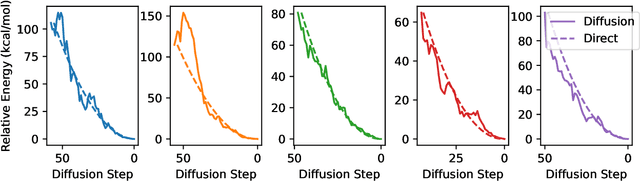
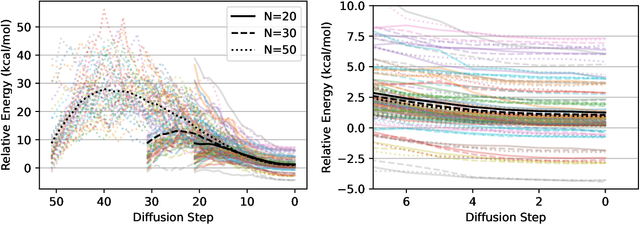
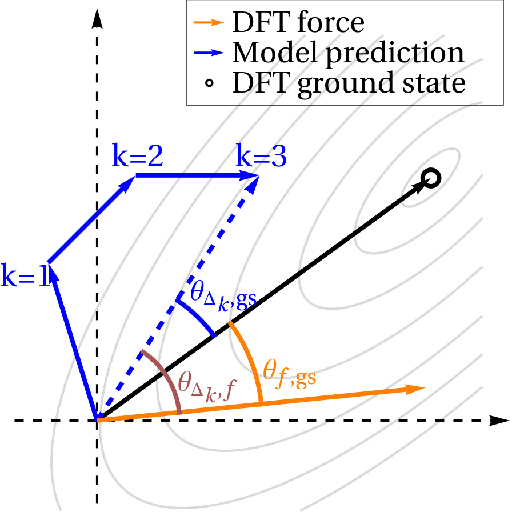
We present an investigation into diffusion models for molecular generation, with the aim of better understanding how their predictions compare to the results of physics-based calculations. The investigation into these models is driven by their potential to significantly accelerate electronic structure calculations using machine learning, without requiring expensive first-principles datasets for training interatomic potentials. We find that the inference process of a popular diffusion model for de novo molecular generation is divided into an exploration phase, where the model chooses the atomic species, and a relaxation phase, where it adjusts the atomic coordinates to find a low-energy geometry. As training proceeds, we show that the model initially learns about the first-order structure of the potential energy surface, and then later learns about higher-order structure. We also find that the relaxation phase of the diffusion model can be re-purposed to sample the Boltzmann distribution over conformations and to carry out structure relaxations. For structure relaxations, the model finds geometries with ~10x lower energy than those produced by a classical force field for small organic molecules. Initializing a density functional theory (DFT) relaxation at the diffusion-produced structures yields a >2x speedup to the DFT relaxation when compared to initializing at structures relaxed with a classical force field.
MOFDiff: Coarse-grained Diffusion for Metal-Organic Framework Design
Oct 16, 2023



Metal-organic frameworks (MOFs) are of immense interest in applications such as gas storage and carbon capture due to their exceptional porosity and tunable chemistry. Their modular nature has enabled the use of template-based methods to generate hypothetical MOFs by combining molecular building blocks in accordance with known network topologies. However, the ability of these methods to identify top-performing MOFs is often hindered by the limited diversity of the resulting chemical space. In this work, we propose MOFDiff: a coarse-grained (CG) diffusion model that generates CG MOF structures through a denoising diffusion process over the coordinates and identities of the building blocks. The all-atom MOF structure is then determined through a novel assembly algorithm. Equivariant graph neural networks are used for the diffusion model to respect the permutational and roto-translational symmetries. We comprehensively evaluate our model's capability to generate valid and novel MOF structures and its effectiveness in designing outstanding MOF materials for carbon capture applications with molecular simulations.
Structured information extraction from complex scientific text with fine-tuned large language models
Dec 10, 2022



Intelligently extracting and linking complex scientific information from unstructured text is a challenging endeavor particularly for those inexperienced with natural language processing. Here, we present a simple sequence-to-sequence approach to joint named entity recognition and relation extraction for complex hierarchical information in scientific text. The approach leverages a pre-trained large language model (LLM), GPT-3, that is fine-tuned on approximately 500 pairs of prompts (inputs) and completions (outputs). Information is extracted either from single sentences or across sentences in abstracts/passages, and the output can be returned as simple English sentences or a more structured format, such as a list of JSON objects. We demonstrate that LLMs trained in this way are capable of accurately extracting useful records of complex scientific knowledge for three representative tasks in materials chemistry: linking dopants with their host materials, cataloging metal-organic frameworks, and general chemistry/phase/morphology/application information extraction. This approach represents a simple, accessible, and highly-flexible route to obtaining large databases of structured knowledge extracted from unstructured text. An online demo is available at http://www.matscholar.com/info-extraction.