 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInvestigating the Behavior of Diffusion Models for Accelerating Electronic Structure Calculations
Nov 02, 2023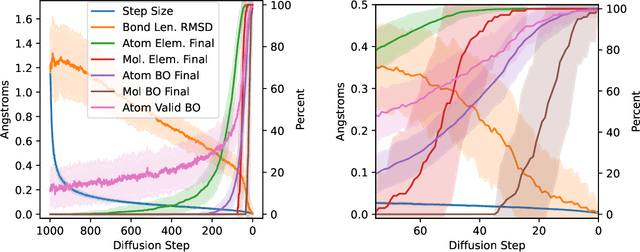
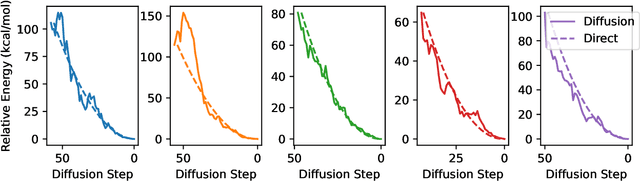
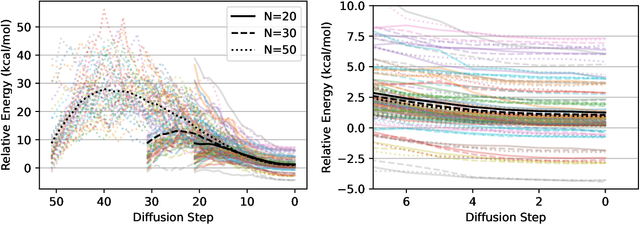
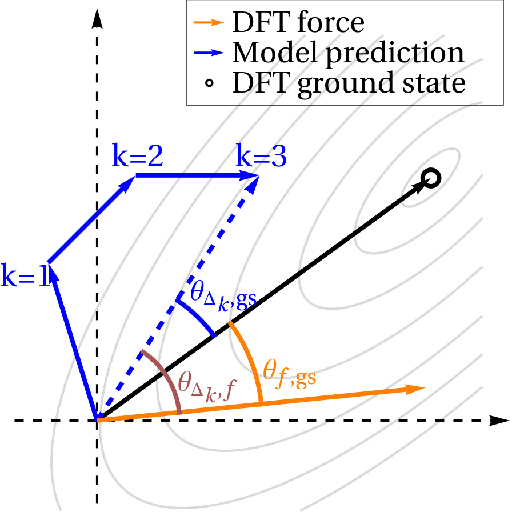
We present an investigation into diffusion models for molecular generation, with the aim of better understanding how their predictions compare to the results of physics-based calculations. The investigation into these models is driven by their potential to significantly accelerate electronic structure calculations using machine learning, without requiring expensive first-principles datasets for training interatomic potentials. We find that the inference process of a popular diffusion model for de novo molecular generation is divided into an exploration phase, where the model chooses the atomic species, and a relaxation phase, where it adjusts the atomic coordinates to find a low-energy geometry. As training proceeds, we show that the model initially learns about the first-order structure of the potential energy surface, and then later learns about higher-order structure. We also find that the relaxation phase of the diffusion model can be re-purposed to sample the Boltzmann distribution over conformations and to carry out structure relaxations. For structure relaxations, the model finds geometries with ~10x lower energy than those produced by a classical force field for small organic molecules. Initializing a density functional theory (DFT) relaxation at the diffusion-produced structures yields a >2x speedup to the DFT relaxation when compared to initializing at structures relaxed with a classical force field.
The Carbon Footprint of Machine Learning Training Will Plateau, Then Shrink
Apr 11, 2022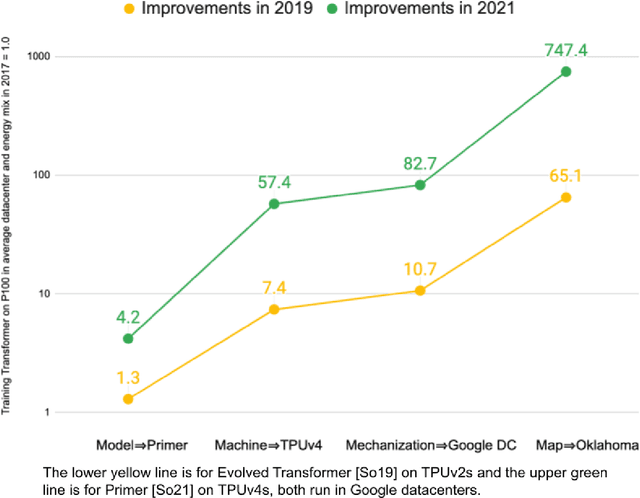
Machine Learning (ML) workloads have rapidly grown in importance, but raised concerns about their carbon footprint. Four best practices can reduce ML training energy by up to 100x and CO2 emissions up to 1000x. By following best practices, overall ML energy use (across research, development, and production) held steady at <15% of Google's total energy use for the past three years. If the whole ML field were to adopt best practices, total carbon emissions from training would reduce. Hence, we recommend that ML papers include emissions explicitly to foster competition on more than just model quality. Estimates of emissions in papers that omitted them have been off 100x-100,000x, so publishing emissions has the added benefit of ensuring accurate accounting. Given the importance of climate change, we must get the numbers right to make certain that we work on its biggest challenges.
C5T5: Controllable Generation of Organic Molecules with Transformers
Aug 23, 2021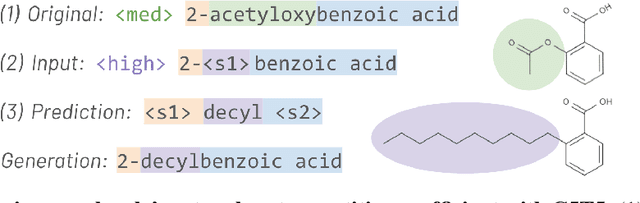

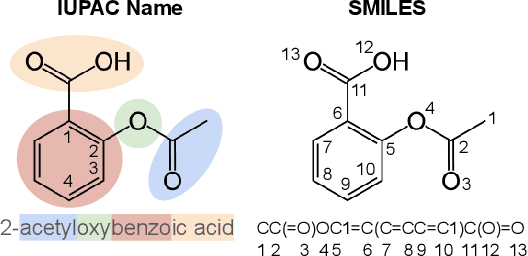
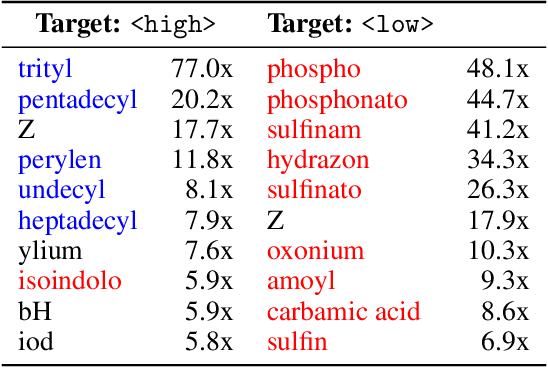
Methods for designing organic materials with desired properties have high potential impact across fields such as medicine, renewable energy, petrochemical engineering, and agriculture. However, using generative modeling to design substances with desired properties is difficult because candidate compounds must satisfy multiple constraints, including synthetic accessibility and other metrics that are intuitive to domain experts but challenging to quantify. We propose C5T5, a novel self-supervised pretraining method that enables transformers to make zero-shot select-and-replace edits, altering organic substances towards desired property values. C5T5 operates on IUPAC names -- a standardized molecular representation that intuitively encodes rich structural information for organic chemists but that has been largely ignored by the ML community. Our technique requires no edited molecule pairs to train and only a rough estimate of molecular properties, and it has the potential to model long-range dependencies and symmetric molecular structures more easily than graph-based methods. C5T5 also provides a powerful interface to domain experts: it grants users fine-grained control over the generative process by selecting and replacing IUPAC name fragments, which enables experts to leverage their intuitions about structure-activity relationships. We demonstrate C5T5's effectiveness on four physical properties relevant for drug discovery, showing that it learns successful and chemically intuitive strategies for altering molecules towards desired property values.
Carbon Emissions and Large Neural Network Training
Apr 23, 2021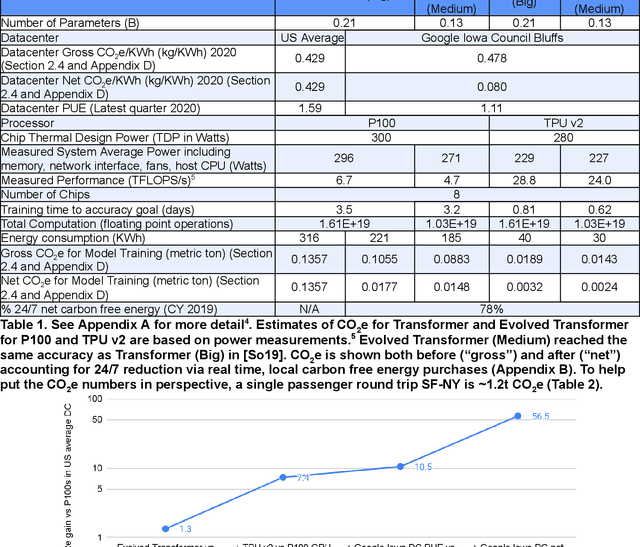
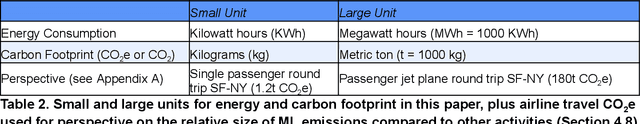
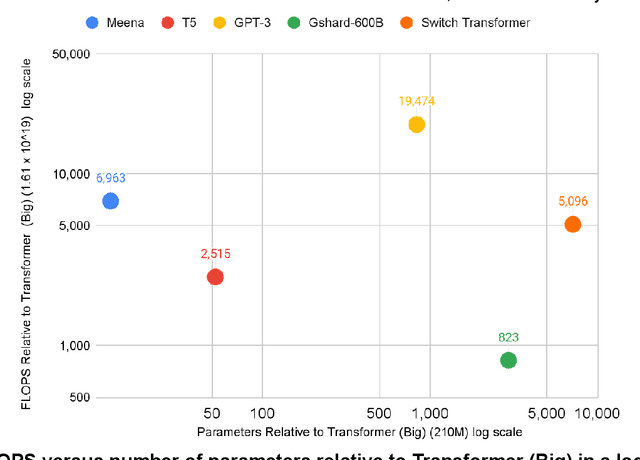
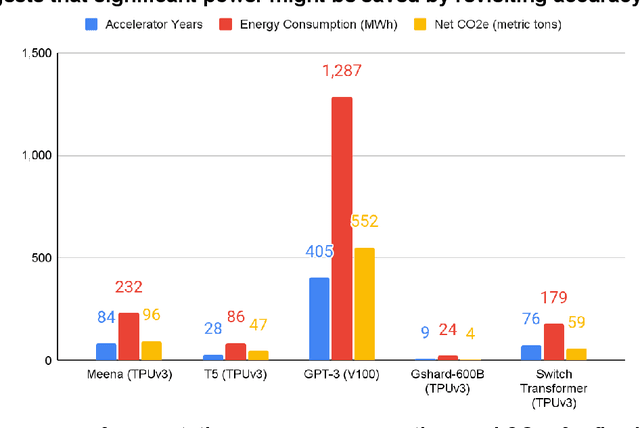
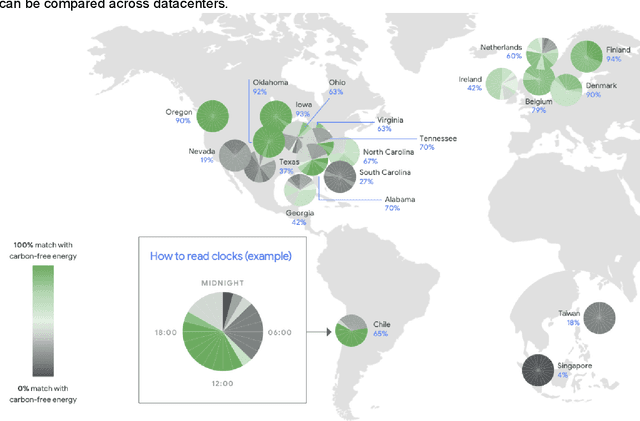
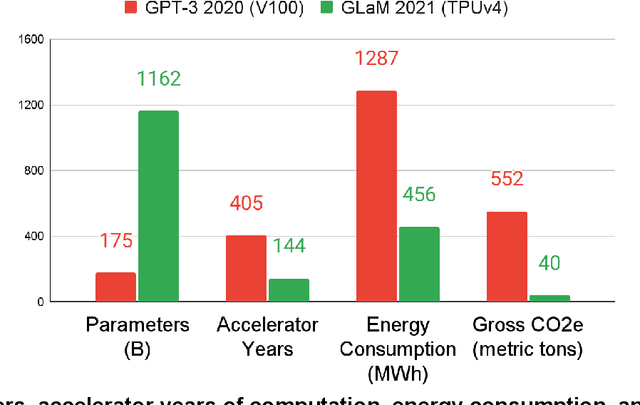
The computation demand for machine learning (ML) has grown rapidly recently, which comes with a number of costs. Estimating the energy cost helps measure its environmental impact and finding greener strategies, yet it is challenging without detailed information. We calculate the energy use and carbon footprint of several recent large models-T5, Meena, GShard, Switch Transformer, and GPT-3-and refine earlier estimates for the neural architecture search that found Evolved Transformer. We highlight the following opportunities to improve energy efficiency and CO2 equivalent emissions (CO2e): Large but sparsely activated DNNs can consume <1/10th the energy of large, dense DNNs without sacrificing accuracy despite using as many or even more parameters. Geographic location matters for ML workload scheduling since the fraction of carbon-free energy and resulting CO2e vary ~5X-10X, even within the same country and the same organization. We are now optimizing where and when large models are trained. Specific datacenter infrastructure matters, as Cloud datacenters can be ~1.4-2X more energy efficient than typical datacenters, and the ML-oriented accelerators inside them can be ~2-5X more effective than off-the-shelf systems. Remarkably, the choice of DNN, datacenter, and processor can reduce the carbon footprint up to ~100-1000X. These large factors also make retroactive estimates of energy cost difficult. To avoid miscalculations, we believe ML papers requiring large computational resources should make energy consumption and CO2e explicit when practical. We are working to be more transparent about energy use and CO2e in our future research. To help reduce the carbon footprint of ML, we believe energy usage and CO2e should be a key metric in evaluating models, and we are collaborating with MLPerf developers to include energy usage during training and inference in this industry standard benchmark.
FetchSGD: Communication-Efficient Federated Learning with Sketching
Jul 15, 2020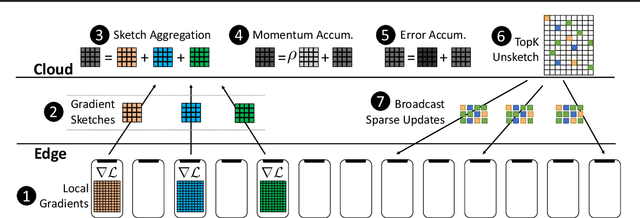

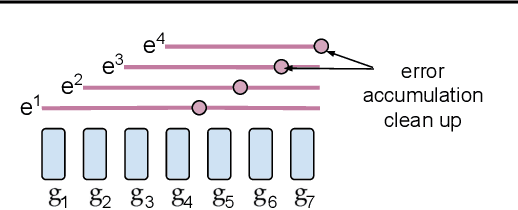
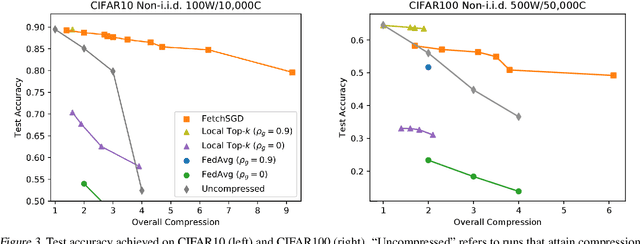
Existing approaches to federated learning suffer from a communication bottleneck as well as convergence issues due to sparse client participation. In this paper we introduce a novel algorithm, called FetchSGD, to overcome these challenges. FetchSGD compresses model updates using a Count Sketch, and then takes advantage of the mergeability of sketches to combine model updates from many workers. A key insight in the design of FetchSGD is that, because the Count Sketch is linear, momentum and error accumulation can both be carried out within the sketch. This allows the algorithm to move momentum and error accumulation from clients to the central aggregator, overcoming the challenges of sparse client participation while still achieving high compression rates and good convergence. We prove that FetchSGD has favorable convergence guarantees, and we demonstrate its empirical effectiveness by training two residual networks and a transformer model.
Communication-efficient distributed SGD with Sketching
Mar 12, 2019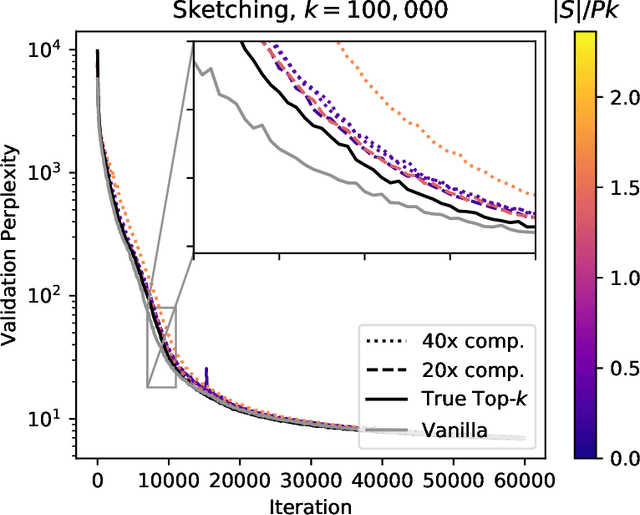
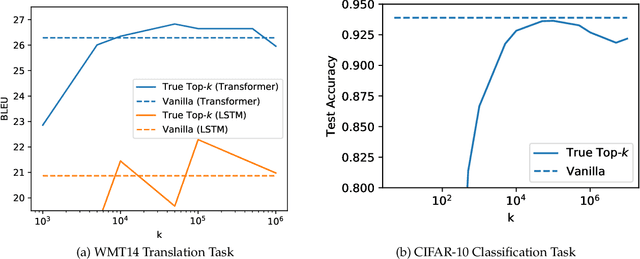
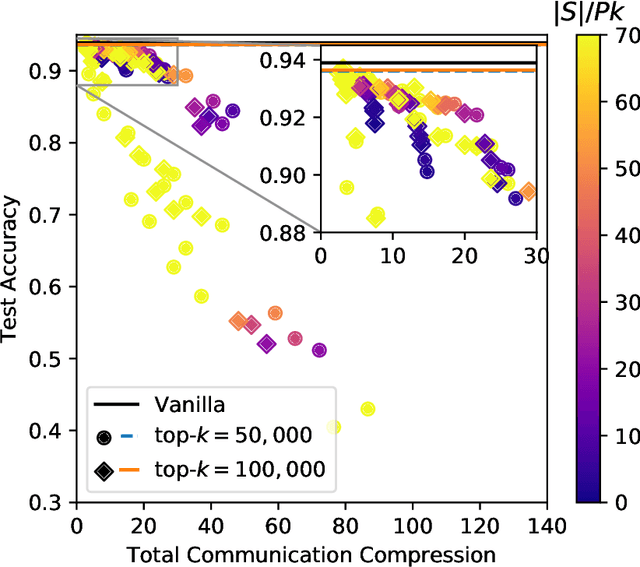
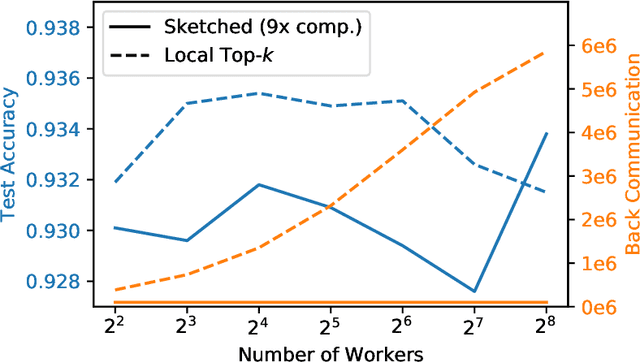
Large-scale distributed training of neural networks is often limited by network bandwidth, wherein the communication time overwhelms the local computation time. Motivated by the success of sketching methods in sub-linear/streaming algorithms, we propose a sketching-based approach to minimize the communication costs between nodes without losing accuracy. In our proposed method, workers in a distributed, synchronous training setting send sketches of their gradient vectors to the parameter server instead of the full gradient vector. Leveraging the theoretical properties of sketches, we show that this method recovers the favorable convergence guarantees of single-machine top-$k$ SGD. Furthermore, when applied to a model with $d$ dimensions on $W$ workers, our method requires only $\Theta(kW)$ bytes of communication, compared to $\Omega(dW)$ for vanilla distributed SGD. To validate our method, we run experiments using a residual network trained on the CIFAR-10 dataset. We achieve no drop in validation accuracy with a compression ratio of 4, or about 1 percentage point drop with a compression ratio of 8. We also demonstrate that our method scales to many workers.