 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHigher-Order Equivariant Neural Networks for Charge Density Prediction in Materials
Dec 08, 2023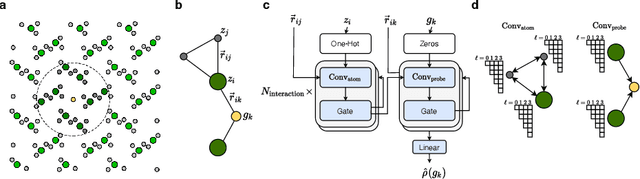

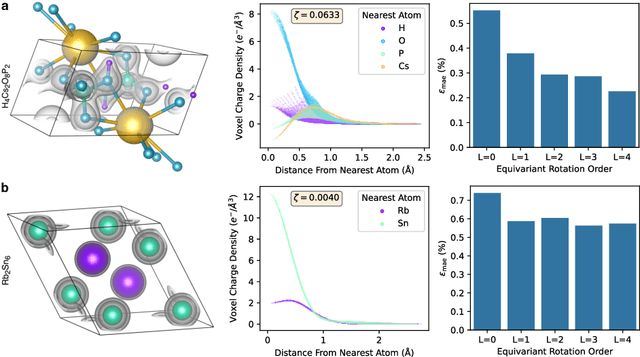
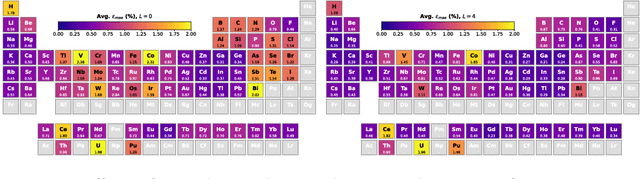
The calculation of electron density distribution using density functional theory (DFT) in materials and molecules is central to the study of their quantum and macro-scale properties, yet accurate and efficient calculation remains a long-standing challenge in the field of material science. This work introduces ChargE3Net, an E(3)-equivariant graph neural network for predicting electron density in atomic systems. ChargE3Net achieves equivariance through the use of higher-order tensor representations, and directly predicts the charge density at any arbitrary point in the system. We show that our method achieves greater performance than prior work on large and diverse sets of molecules and materials, and scales to larger systems than what is feasible to compute with DFT. Using predicted electron densities as an initialization, we show that fewer self-consistent iterations are required to converge DFT over the default initialization. In addition, we show that non-self-consistent calculations using the predicted electron densities can predict electronic and thermodynamic properties of materials at near-DFT accuracy.
Investigating the Behavior of Diffusion Models for Accelerating Electronic Structure Calculations
Nov 02, 2023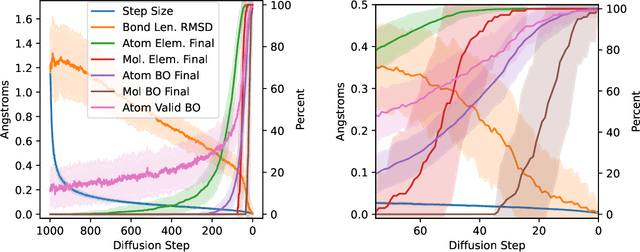
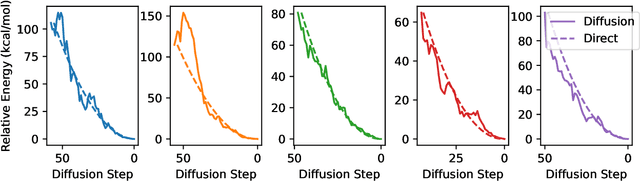
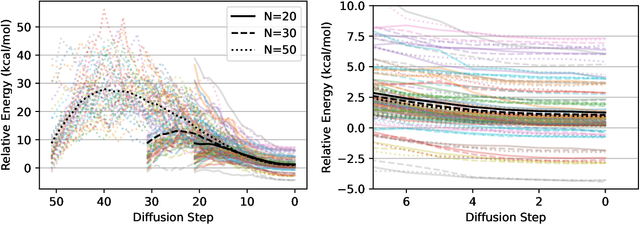
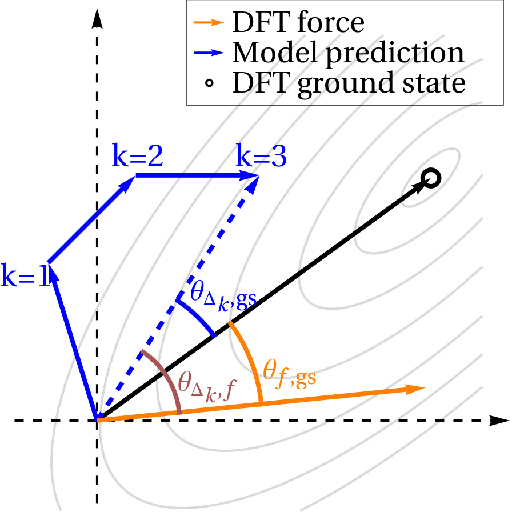
We present an investigation into diffusion models for molecular generation, with the aim of better understanding how their predictions compare to the results of physics-based calculations. The investigation into these models is driven by their potential to significantly accelerate electronic structure calculations using machine learning, without requiring expensive first-principles datasets for training interatomic potentials. We find that the inference process of a popular diffusion model for de novo molecular generation is divided into an exploration phase, where the model chooses the atomic species, and a relaxation phase, where it adjusts the atomic coordinates to find a low-energy geometry. As training proceeds, we show that the model initially learns about the first-order structure of the potential energy surface, and then later learns about higher-order structure. We also find that the relaxation phase of the diffusion model can be re-purposed to sample the Boltzmann distribution over conformations and to carry out structure relaxations. For structure relaxations, the model finds geometries with ~10x lower energy than those produced by a classical force field for small organic molecules. Initializing a density functional theory (DFT) relaxation at the diffusion-produced structures yields a >2x speedup to the DFT relaxation when compared to initializing at structures relaxed with a classical force field.