 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHigher-Order Equivariant Neural Networks for Charge Density Prediction in Materials
Paper and Code
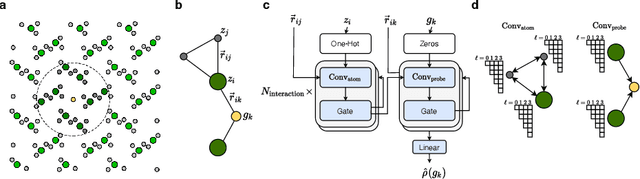

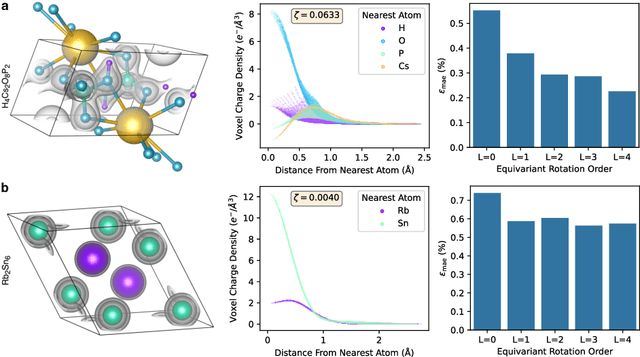
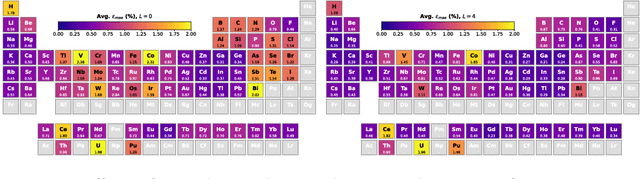
The calculation of electron density distribution using density functional theory (DFT) in materials and molecules is central to the study of their quantum and macro-scale properties, yet accurate and efficient calculation remains a long-standing challenge in the field of material science. This work introduces ChargE3Net, an E(3)-equivariant graph neural network for predicting electron density in atomic systems. ChargE3Net achieves equivariance through the use of higher-order tensor representations, and directly predicts the charge density at any arbitrary point in the system. We show that our method achieves greater performance than prior work on large and diverse sets of molecules and materials, and scales to larger systems than what is feasible to compute with DFT. Using predicted electron densities as an initialization, we show that fewer self-consistent iterations are required to converge DFT over the default initialization. In addition, we show that non-self-consistent calculations using the predicted electron densities can predict electronic and thermodynamic properties of materials at near-DFT accuracy.