 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOvercoming systematic softening in universal machine learning interatomic potentials by fine-tuning
May 11, 2024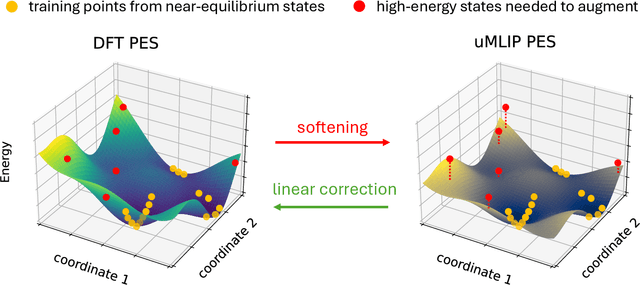
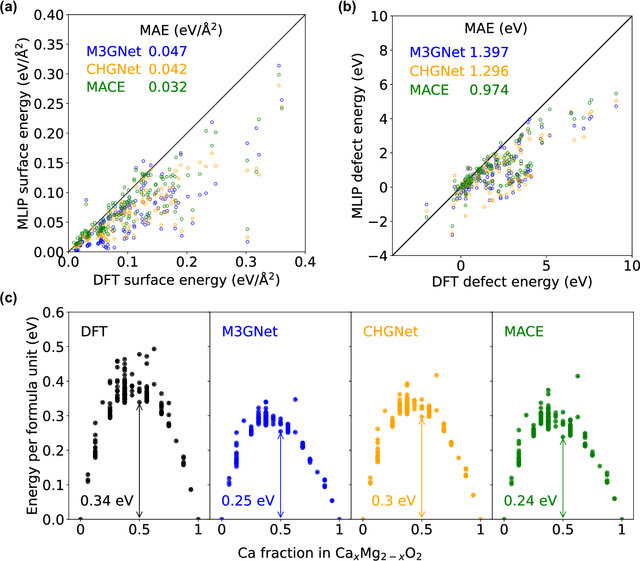
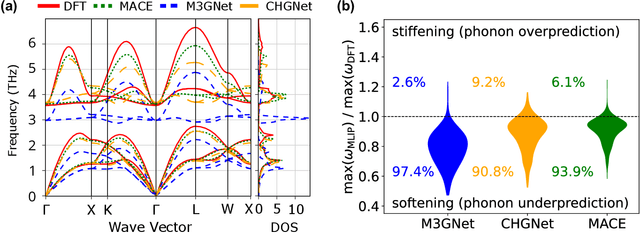
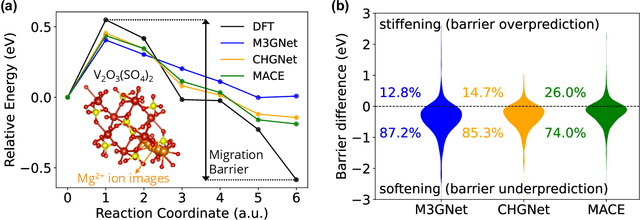
Machine learning interatomic potentials (MLIPs) have introduced a new paradigm for atomic simulations. Recent advancements have seen the emergence of universal MLIPs (uMLIPs) that are pre-trained on diverse materials datasets, providing opportunities for both ready-to-use universal force fields and robust foundations for downstream machine learning refinements. However, their performance in extrapolating to out-of-distribution complex atomic environments remains unclear. In this study, we highlight a consistent potential energy surface (PES) softening effect in three uMLIPs: M3GNet, CHGNet, and MACE-MP-0, which is characterized by energy and force under-prediction in a series of atomic-modeling benchmarks including surfaces, defects, solid-solution energetics, phonon vibration modes, ion migration barriers, and general high-energy states. We find that the PES softening behavior originates from a systematic underprediction error of the PES curvature, which derives from the biased sampling of near-equilibrium atomic arrangements in uMLIP pre-training datasets. We demonstrate that the PES softening issue can be effectively rectified by fine-tuning with a single additional data point. Our findings suggest that a considerable fraction of uMLIP errors are highly systematic, and can therefore be efficiently corrected. This result rationalizes the data-efficient fine-tuning performance boost commonly observed with foundational MLIPs. We argue for the importance of a comprehensive materials dataset with improved PES sampling for next-generation foundational MLIPs.
LLaMP: Large Language Model Made Powerful for High-fidelity Materials Knowledge Retrieval and Distillation
Jan 30, 2024



Reducing hallucination of Large Language Models (LLMs) is imperative for use in the sciences where reproducibility is crucial. However, LLMs inherently lack long-term memory, making it a nontrivial, ad hoc, and inevitably biased task to fine-tune them on domain-specific literature and data. Here we introduce LLaMP, a multimodal retrieval-augmented generation (RAG) framework of multiple data-aware reasoning-and-acting (ReAct) agents that dynamically interact with computational and experimental data on Materials Project (MP). Without fine-tuning, LLaMP demonstrates an ability to comprehend and integrate various modalities of materials science concepts, fetch relevant data stores on the fly, process higher-order data (such as crystal structures and elastic tensors), and summarize multi-step procedures for solid-state synthesis. We show that LLaMP effectively corrects errors in GPT-3.5's intrinsic knowledge, reducing a 5.21% MAPE on frequently-documented bandgaps and a significant 1103.54% MAPE on formation energies -- errors that GPT-3.5 seems to derive from mixed data sources. Additionally, LLaMP substantially reduces the hallucinated volumetric strain in a diamond cubic silicon structure from 66.3% to 0. The proposed framework offers an intuitive and nearly hallucination-free approach to exploring materials informatics and establishes a pathway for knowledge distillation and fine-tuning other language models. We envision the framework as a valuable component for scientific hypotheses and a foundation for future autonomous laboratories where multiple LLM agents communicate and cooperate with robotics to drive material synthesis and chemical reactions without hard-coded human logic and intervention.
Pushing the Pareto front of band gap and permittivity: ML-guided search for dielectric materials
Jan 11, 2024



Materials with high-dielectric constant easily polarize under external electric fields, allowing them to perform essential functions in many modern electronic devices. Their practical utility is determined by two conflicting properties: high dielectric constants tend to occur in materials with narrow band gaps, limiting the operating voltage before dielectric breakdown. We present a high-throughput workflow that combines element substitution, ML pre-screening, ab initio simulation and human expert intuition to efficiently explore the vast space of unknown materials for potential dielectrics, leading to the synthesis and characterization of two novel dielectric materials, CsTaTeO6 and Bi2Zr2O7. Our key idea is to deploy ML in a multi-objective optimization setting with concave Pareto front. While usually considered more challenging than single-objective optimization, we argue and show preliminary evidence that the $1/x$-correlation between band gap and permittivity in fact makes the task more amenable to ML methods by allowing separate models for band gap and permittivity to each operate in regions of good training support while still predicting materials of exceptional merit. To our knowledge, this is the first instance of successful ML-guided multi-objective materials optimization achieving experimental synthesis and characterization. CsTaTeO6 is a structure generated via element substitution not present in our reference data sources, thus exemplifying successful de-novo materials design. Meanwhile, we report the first high-purity synthesis and dielectric characterization of Bi2Zr2O7 with a band gap of 2.27 eV and a permittivity of 20.5, meeting all target metrics of our multi-objective search.
Matbench Discovery -- An evaluation framework for machine learning crystal stability prediction
Aug 28, 2023Matbench Discovery simulates the deployment of machine learning (ML) energy models in a high-throughput search for stable inorganic crystals. We address the disconnect between (i) thermodynamic stability and formation energy and (ii) in-domain vs out-of-distribution performance. Alongside this paper, we publish a Python package to aid with future model submissions and a growing online leaderboard with further insights into trade-offs between various performance metrics. To answer the question which ML methodology performs best at materials discovery, our initial release explores a variety of models including random forests, graph neural networks (GNN), one-shot predictors, iterative Bayesian optimizers and universal interatomic potentials (UIP). Ranked best-to-worst by their test set F1 score on thermodynamic stability prediction, we find CHGNet > M3GNet > MACE > ALIGNN > MEGNet > CGCNN > CGCNN+P > Wrenformer > BOWSR > Voronoi tessellation fingerprints with random forest. The top 3 models are UIPs, the winning methodology for ML-guided materials discovery, achieving F1 scores of ~0.6 for crystal stability classification and discovery acceleration factors (DAF) of up to 5x on the first 10k most stable predictions compared to dummy selection from our test set. We also highlight a sharp disconnect between commonly used global regression metrics and more task-relevant classification metrics. Accurate regressors are susceptible to unexpectedly high false-positive rates if those accurate predictions lie close to the decision boundary at 0 eV/atom above the convex hull where most materials are. Our results highlight the need to focus on classification metrics that actually correlate with improved stability hit rate.