 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMatbench Discovery -- An evaluation framework for machine learning crystal stability prediction
Aug 28, 2023Matbench Discovery simulates the deployment of machine learning (ML) energy models in a high-throughput search for stable inorganic crystals. We address the disconnect between (i) thermodynamic stability and formation energy and (ii) in-domain vs out-of-distribution performance. Alongside this paper, we publish a Python package to aid with future model submissions and a growing online leaderboard with further insights into trade-offs between various performance metrics. To answer the question which ML methodology performs best at materials discovery, our initial release explores a variety of models including random forests, graph neural networks (GNN), one-shot predictors, iterative Bayesian optimizers and universal interatomic potentials (UIP). Ranked best-to-worst by their test set F1 score on thermodynamic stability prediction, we find CHGNet > M3GNet > MACE > ALIGNN > MEGNet > CGCNN > CGCNN+P > Wrenformer > BOWSR > Voronoi tessellation fingerprints with random forest. The top 3 models are UIPs, the winning methodology for ML-guided materials discovery, achieving F1 scores of ~0.6 for crystal stability classification and discovery acceleration factors (DAF) of up to 5x on the first 10k most stable predictions compared to dummy selection from our test set. We also highlight a sharp disconnect between commonly used global regression metrics and more task-relevant classification metrics. Accurate regressors are susceptible to unexpectedly high false-positive rates if those accurate predictions lie close to the decision boundary at 0 eV/atom above the convex hull where most materials are. Our results highlight the need to focus on classification metrics that actually correlate with improved stability hit rate.
Predicting materials properties without crystal structure: Deep representation learning from stoichiometry
Oct 29, 2019
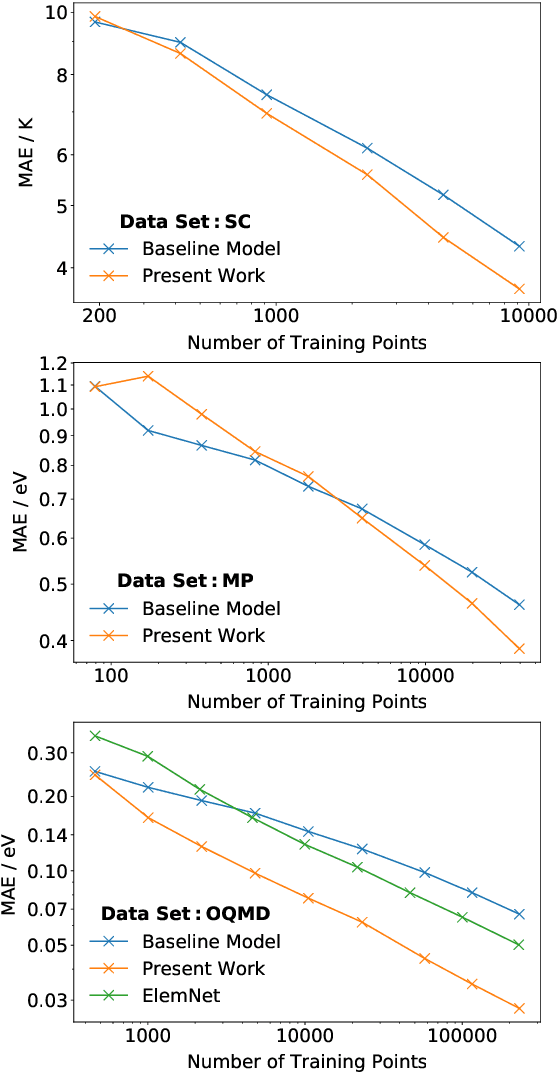
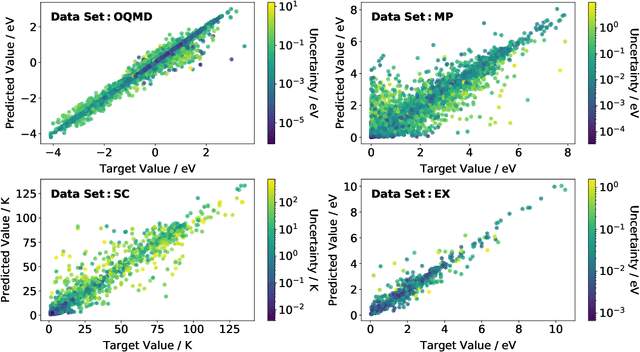
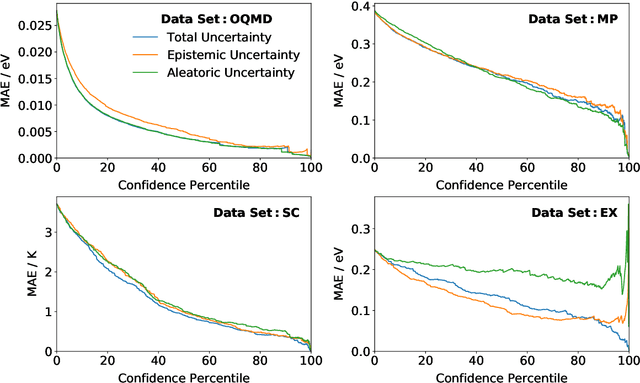
Machine learning can accelerate materials discovery by accurately predicting materials properties with low computational cost. However, the model inputs remain a key stumbling block: current methods typically use hand-engineered descriptors constructed from knowledge of either the full crystal structure -- applicable only to materials with experimentally measured structures as crystal structure prediction is computationally expensive -- or the stoichiometry. We develop a machine learning approach that takes only the stoichiometry as input and automatically learns appropriate and systematically improvable descriptors from data. Our key insight is to treat the stoichiometric formula as a dense weighted graph between elements. Compared to the state of the art, our approach achieves lower error on a plethora of challenging material properties. Moreover, our model can estimate its own uncertainty as well as transfer its learnt representation, extracting useful information from a cognate data-abundant task to deploy on a data-poor task.