 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTransport Novelty Distance: A Distributional Metric for Evaluating Material Generative Models
Dec 10, 2025



Recent advances in generative machine learning have opened new possibilities for the discovery and design of novel materials. However, as these models become more sophisticated, the need for rigorous and meaningful evaluation metrics has grown. Existing evaluation approaches often fail to capture both the quality and novelty of generated structures, limiting our ability to assess true generative performance. In this paper, we introduce the Transport Novelty Distance (TNovD) to judge generative models used for materials discovery jointly by the quality and novelty of the generated materials. Based on ideas from Optimal Transport theory, TNovD uses a coupling between the features of the training and generated sets, which is refined into a quality and memorization regime by a threshold. The features are generated from crystal structures using a graph neural network that is trained to distinguish between materials, their augmented counterparts, and differently sized supercells using contrastive learning. We evaluate our proposed metric on typical toy experiments relevant for crystal structure prediction, including memorization, noise injection and lattice deformations. Additionally, we validate the TNovD on the MP20 validation set and the WBM substitution dataset, demonstrating that it is capable of detecting both memorized and low-quality material data. We also benchmark the performance of several popular material generative models. While introduced for materials, our TNovD framework is domain-agnostic and can be adapted for other areas, such as images and molecules.
SynCoTrain: A Dual Classifier PU-learning Framework for Synthesizability Prediction
Nov 18, 2024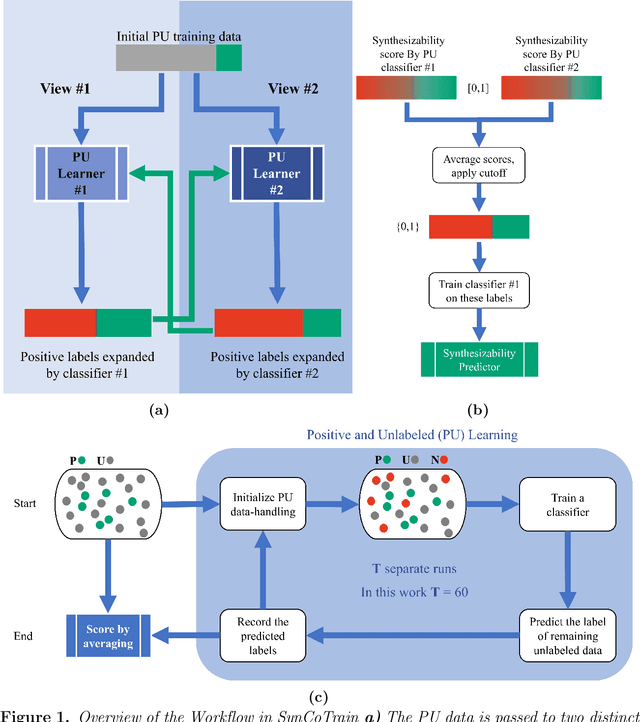

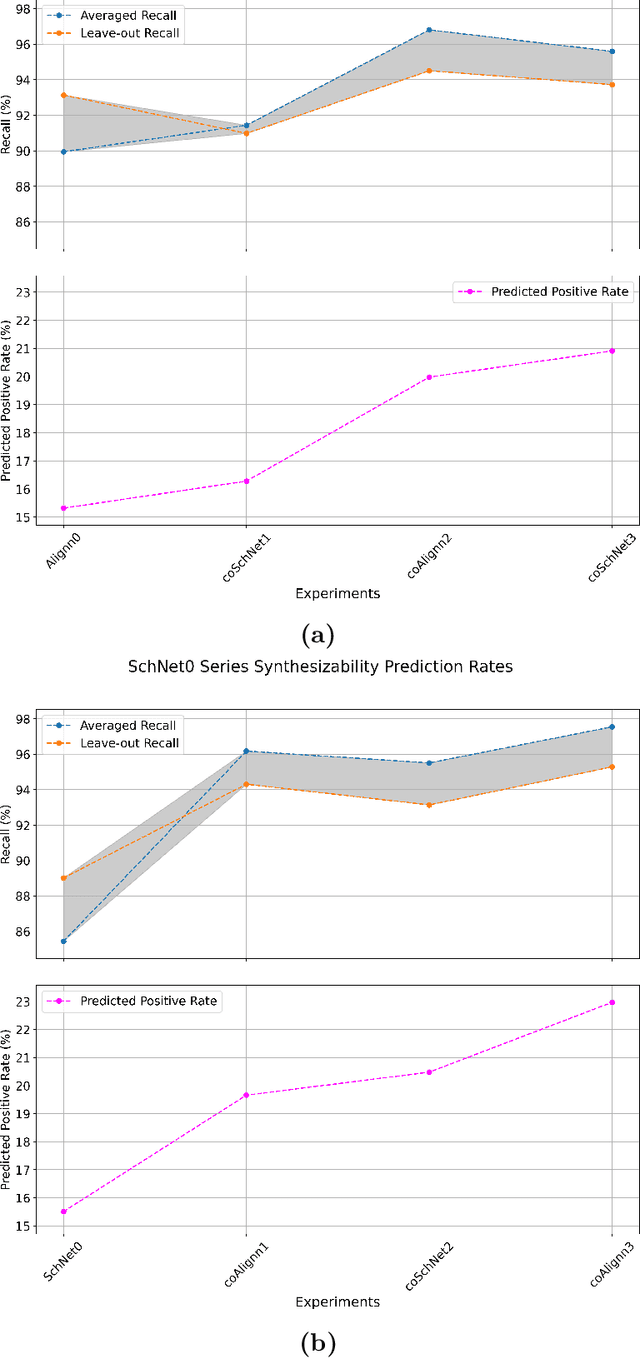
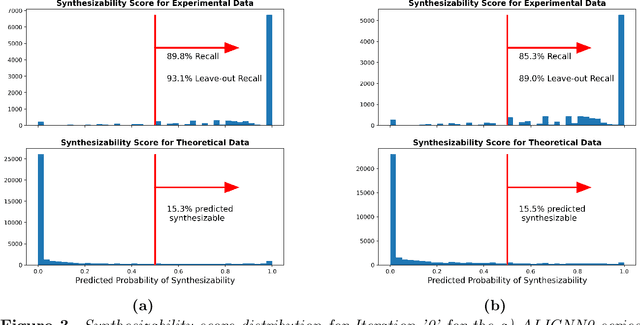
Material discovery is a cornerstone of modern science, driving advancements in diverse disciplines from biomedical technology to climate solutions. Predicting synthesizability, a critical factor in realizing novel materials, remains a complex challenge due to the limitations of traditional heuristics and thermodynamic proxies. While stability metrics such as formation energy offer partial insights, they fail to account for kinetic factors and technological constraints that influence synthesis outcomes. These challenges are further compounded by the scarcity of negative data, as failed synthesis attempts are often unpublished or context-specific. We present SynCoTrain, a semi-supervised machine learning model designed to predict the synthesizability of materials. SynCoTrain employs a co-training framework leveraging two complementary graph convolutional neural networks: SchNet and ALIGNN. By iteratively exchanging predictions between classifiers, SynCoTrain mitigates model bias and enhances generalizability. Our approach uses Positive and Unlabeled (PU) Learning to address the absence of explicit negative data, iteratively refining predictions through collaborative learning. The model demonstrates robust performance, achieving high recall on internal and leave-out test sets. By focusing on oxide crystals, a well-characterized material family with extensive experimental data, we establish SynCoTrain as a reliable tool for predicting synthesizability while balancing dataset variability and computational efficiency. This work highlights the potential of co-training to advance high-throughput materials discovery and generative research, offering a scalable solution to the challenge of synthesizability prediction.
Matbench Discovery -- An evaluation framework for machine learning crystal stability prediction
Aug 28, 2023Matbench Discovery simulates the deployment of machine learning (ML) energy models in a high-throughput search for stable inorganic crystals. We address the disconnect between (i) thermodynamic stability and formation energy and (ii) in-domain vs out-of-distribution performance. Alongside this paper, we publish a Python package to aid with future model submissions and a growing online leaderboard with further insights into trade-offs between various performance metrics. To answer the question which ML methodology performs best at materials discovery, our initial release explores a variety of models including random forests, graph neural networks (GNN), one-shot predictors, iterative Bayesian optimizers and universal interatomic potentials (UIP). Ranked best-to-worst by their test set F1 score on thermodynamic stability prediction, we find CHGNet > M3GNet > MACE > ALIGNN > MEGNet > CGCNN > CGCNN+P > Wrenformer > BOWSR > Voronoi tessellation fingerprints with random forest. The top 3 models are UIPs, the winning methodology for ML-guided materials discovery, achieving F1 scores of ~0.6 for crystal stability classification and discovery acceleration factors (DAF) of up to 5x on the first 10k most stable predictions compared to dummy selection from our test set. We also highlight a sharp disconnect between commonly used global regression metrics and more task-relevant classification metrics. Accurate regressors are susceptible to unexpectedly high false-positive rates if those accurate predictions lie close to the decision boundary at 0 eV/atom above the convex hull where most materials are. Our results highlight the need to focus on classification metrics that actually correlate with improved stability hit rate.