 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePerspective: Towards sustainable exploration of chemical spaces with machine learning
Mar 31, 2026Artificial intelligence is transforming molecular and materials science, but its growing computational and data demands raise critical sustainability challenges. In this Perspective, we examine resource considerations across the AI-driven discovery pipeline--from quantum-mechanical (QM) data generation and model training to automated, self-driving research workflows--building on discussions from the ``SusML workshop: Towards sustainable exploration of chemical spaces with machine learning'' held in Dresden, Germany. In this context, the availability of large quantum datasets has enabled rigorous benchmarking and rapid methodological progress, while also incurring substantial energy and infrastructure costs. We highlight emerging strategies to enhance efficiency, including general-purpose machine learning (ML) models, multi-fidelity approaches, model distillation, and active learning. Moreover, incorporating physics-based constraints within hierarchical workflows, where fast ML surrogates are applied broadly and high-accuracy QM methods are used selectively, can further optimize resource use without compromising reliability. Equally important is bridging the gap between idealized computational predictions and real-world conditions by accounting for synthesizability and multi-objective design criteria, which is essential for practical impact. Finally, we argue that sustainable progress will rely on open data and models, reusable workflows, and domain-specific AI systems that maximize scientific value per unit of computation, enabling efficient and responsible discovery of technological materials and therapeutics.
Transport Novelty Distance: A Distributional Metric for Evaluating Material Generative Models
Dec 10, 2025



Recent advances in generative machine learning have opened new possibilities for the discovery and design of novel materials. However, as these models become more sophisticated, the need for rigorous and meaningful evaluation metrics has grown. Existing evaluation approaches often fail to capture both the quality and novelty of generated structures, limiting our ability to assess true generative performance. In this paper, we introduce the Transport Novelty Distance (TNovD) to judge generative models used for materials discovery jointly by the quality and novelty of the generated materials. Based on ideas from Optimal Transport theory, TNovD uses a coupling between the features of the training and generated sets, which is refined into a quality and memorization regime by a threshold. The features are generated from crystal structures using a graph neural network that is trained to distinguish between materials, their augmented counterparts, and differently sized supercells using contrastive learning. We evaluate our proposed metric on typical toy experiments relevant for crystal structure prediction, including memorization, noise injection and lattice deformations. Additionally, we validate the TNovD on the MP20 validation set and the WBM substitution dataset, demonstrating that it is capable of detecting both memorized and low-quality material data. We also benchmark the performance of several popular material generative models. While introduced for materials, our TNovD framework is domain-agnostic and can be adapted for other areas, such as images and molecules.
34 Examples of LLM Applications in Materials Science and Chemistry: Towards Automation, Assistants, Agents, and Accelerated Scientific Discovery
May 05, 2025
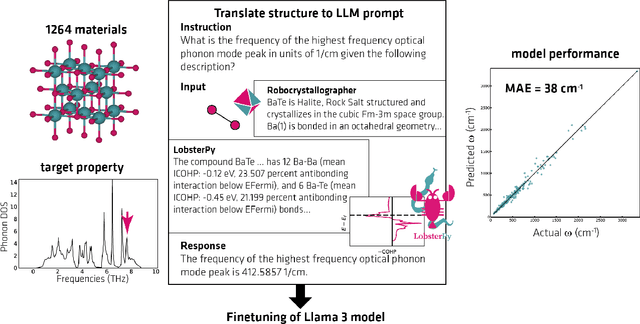
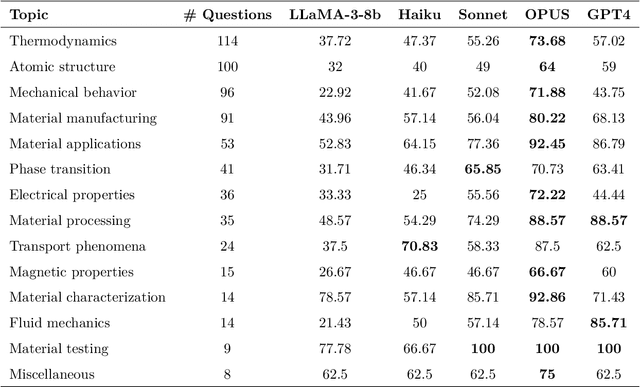
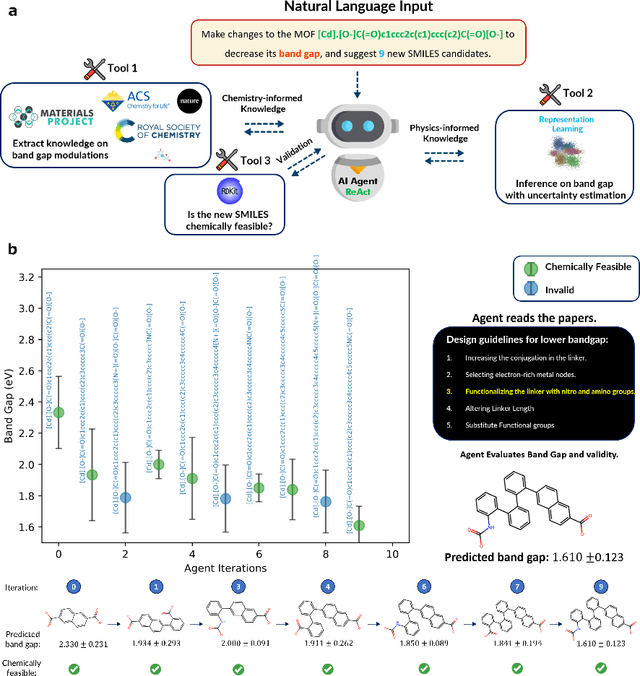
Large Language Models (LLMs) are reshaping many aspects of materials science and chemistry research, enabling advances in molecular property prediction, materials design, scientific automation, knowledge extraction, and more. Recent developments demonstrate that the latest class of models are able to integrate structured and unstructured data, assist in hypothesis generation, and streamline research workflows. To explore the frontier of LLM capabilities across the research lifecycle, we review applications of LLMs through 34 total projects developed during the second annual Large Language Model Hackathon for Applications in Materials Science and Chemistry, a global hybrid event. These projects spanned seven key research areas: (1) molecular and material property prediction, (2) molecular and material design, (3) automation and novel interfaces, (4) scientific communication and education, (5) research data management and automation, (6) hypothesis generation and evaluation, and (7) knowledge extraction and reasoning from the scientific literature. Collectively, these applications illustrate how LLMs serve as versatile predictive models, platforms for rapid prototyping of domain-specific tools, and much more. In particular, improvements in both open source and proprietary LLM performance through the addition of reasoning, additional training data, and new techniques have expanded effectiveness, particularly in low-data environments and interdisciplinary research. As LLMs continue to improve, their integration into scientific workflows presents both new opportunities and new challenges, requiring ongoing exploration, continued refinement, and further research to address reliability, interpretability, and reproducibility.
Reflections from the 2024 Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry
Nov 20, 2024
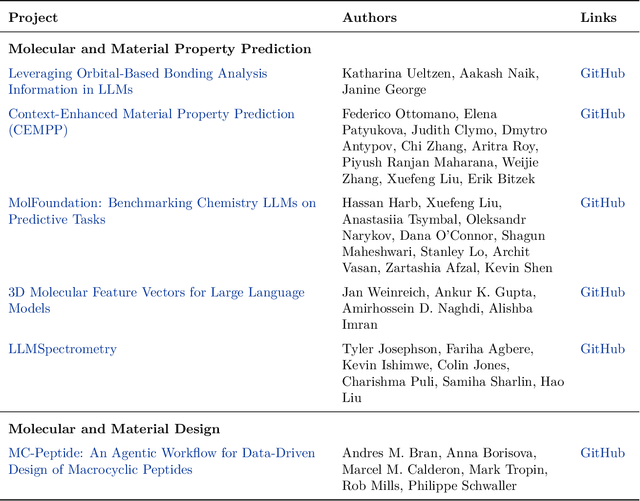
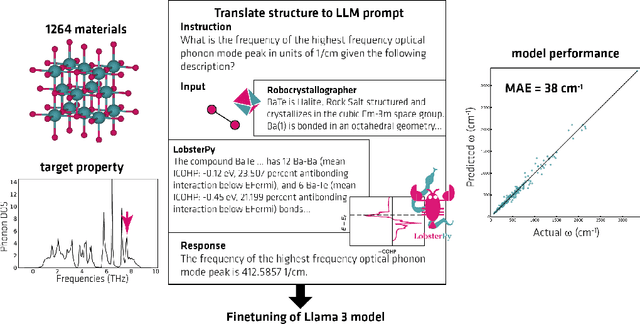
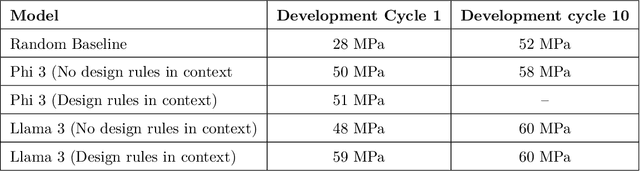
Here, we present the outcomes from the second Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry, which engaged participants across global hybrid locations, resulting in 34 team submissions. The submissions spanned seven key application areas and demonstrated the diverse utility of LLMs for applications in (1) molecular and material property prediction; (2) molecular and material design; (3) automation and novel interfaces; (4) scientific communication and education; (5) research data management and automation; (6) hypothesis generation and evaluation; and (7) knowledge extraction and reasoning from scientific literature. Each team submission is presented in a summary table with links to the code and as brief papers in the appendix. Beyond team results, we discuss the hackathon event and its hybrid format, which included physical hubs in Toronto, Montreal, San Francisco, Berlin, Lausanne, and Tokyo, alongside a global online hub to enable local and virtual collaboration. Overall, the event highlighted significant improvements in LLM capabilities since the previous year's hackathon, suggesting continued expansion of LLMs for applications in materials science and chemistry research. These outcomes demonstrate the dual utility of LLMs as both multipurpose models for diverse machine learning tasks and platforms for rapid prototyping custom applications in scientific research.
SynCoTrain: A Dual Classifier PU-learning Framework for Synthesizability Prediction
Nov 18, 2024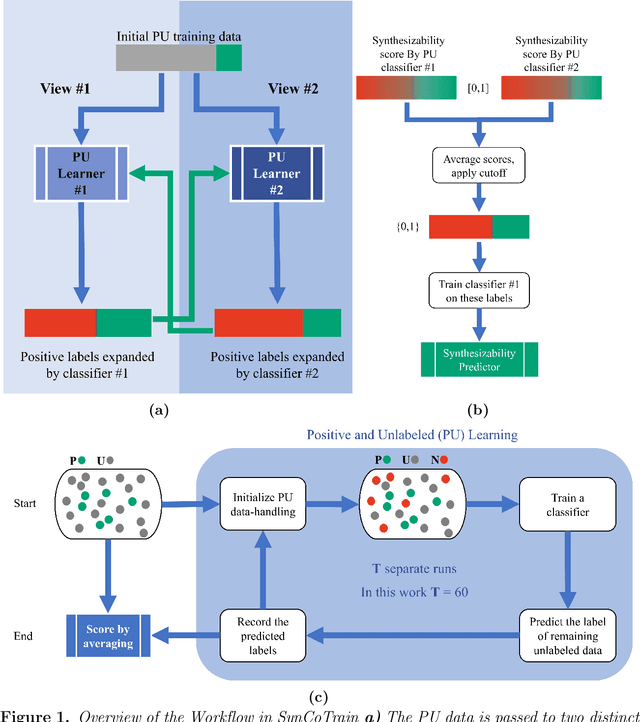

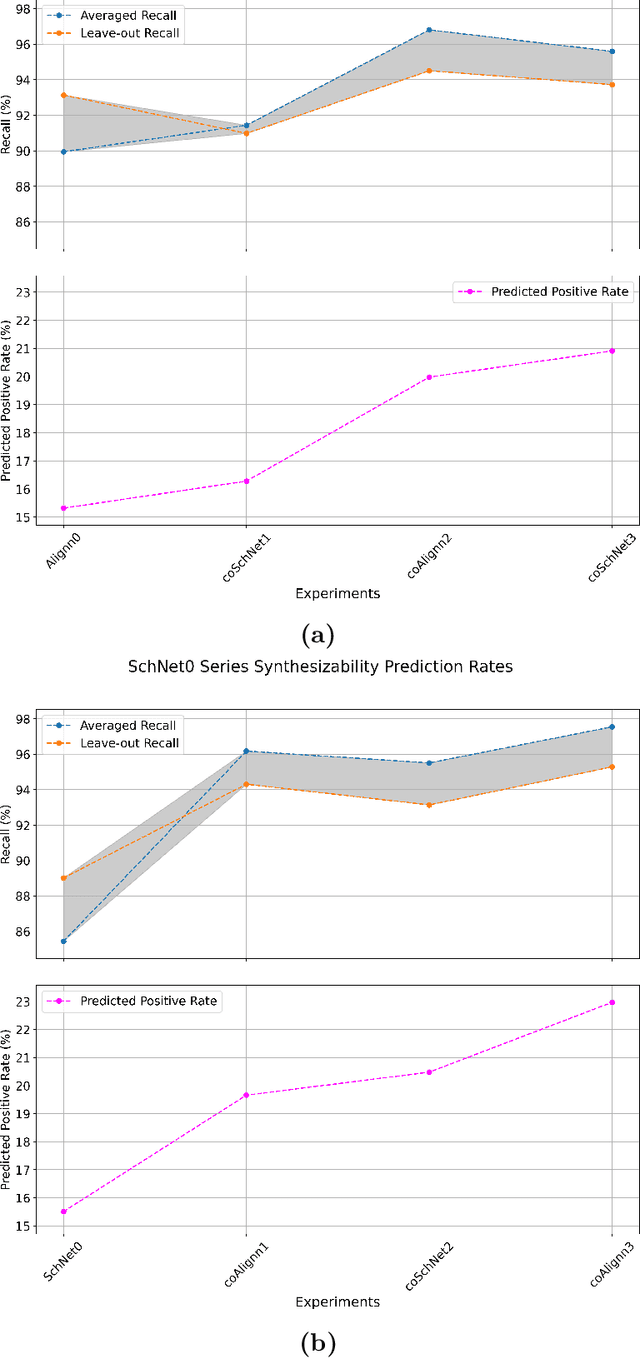
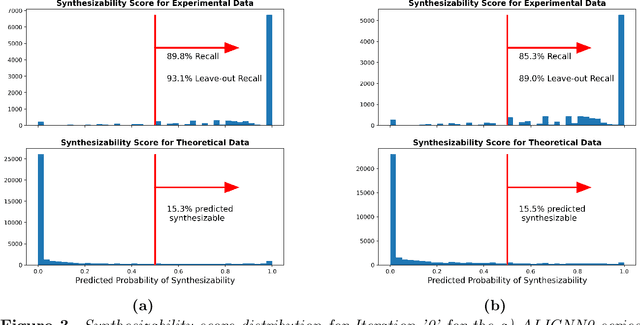
Material discovery is a cornerstone of modern science, driving advancements in diverse disciplines from biomedical technology to climate solutions. Predicting synthesizability, a critical factor in realizing novel materials, remains a complex challenge due to the limitations of traditional heuristics and thermodynamic proxies. While stability metrics such as formation energy offer partial insights, they fail to account for kinetic factors and technological constraints that influence synthesis outcomes. These challenges are further compounded by the scarcity of negative data, as failed synthesis attempts are often unpublished or context-specific. We present SynCoTrain, a semi-supervised machine learning model designed to predict the synthesizability of materials. SynCoTrain employs a co-training framework leveraging two complementary graph convolutional neural networks: SchNet and ALIGNN. By iteratively exchanging predictions between classifiers, SynCoTrain mitigates model bias and enhances generalizability. Our approach uses Positive and Unlabeled (PU) Learning to address the absence of explicit negative data, iteratively refining predictions through collaborative learning. The model demonstrates robust performance, achieving high recall on internal and leave-out test sets. By focusing on oxide crystals, a well-characterized material family with extensive experimental data, we establish SynCoTrain as a reliable tool for predicting synthesizability while balancing dataset variability and computational efficiency. This work highlights the potential of co-training to advance high-throughput materials discovery and generative research, offering a scalable solution to the challenge of synthesizability prediction.