 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMACS: Multi-Agent Reinforcement Learning for Optimization of Crystal Structures
Jun 04, 2025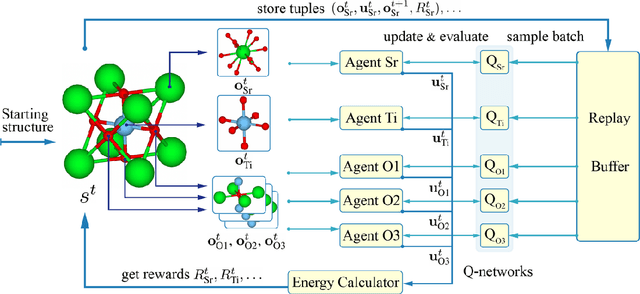



Geometry optimization of atomic structures is a common and crucial task in computational chemistry and materials design. Following the learning to optimize paradigm, we propose a new multi-agent reinforcement learning method called Multi-Agent Crystal Structure optimization (MACS) to address periodic crystal structure optimization. MACS treats geometry optimization as a partially observable Markov game in which atoms are agents that adjust their positions to collectively discover a stable configuration. We train MACS across various compositions of reported crystalline materials to obtain a policy that successfully optimizes structures from the training compositions as well as structures of larger sizes and unseen compositions, confirming its excellent scalability and zero-shot transferability. We benchmark our approach against a broad range of state-of-the-art optimization methods and demonstrate that MACS optimizes periodic crystal structures significantly faster, with fewer energy calculations, and the lowest failure rate.
Reflections from the 2024 Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry
Nov 20, 2024
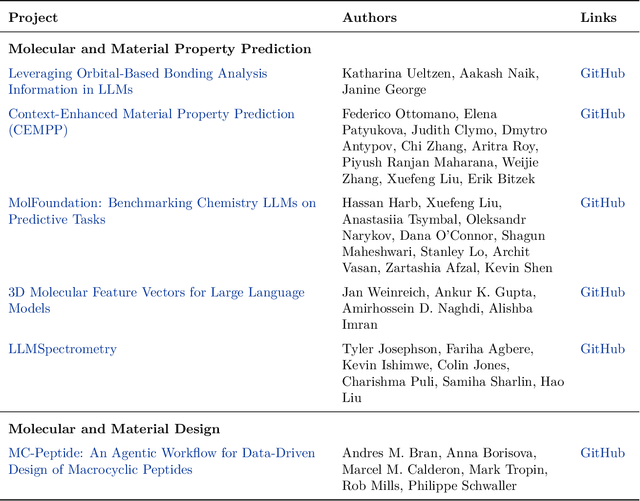
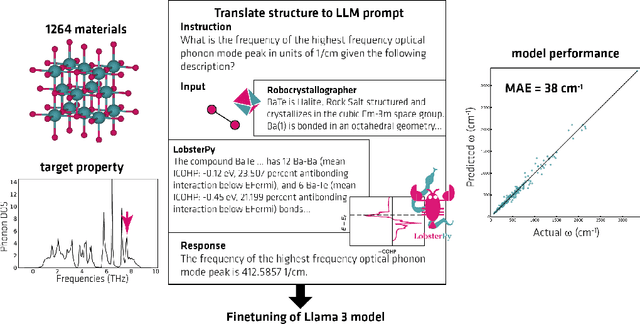
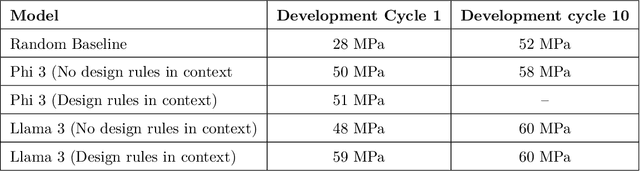
Here, we present the outcomes from the second Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry, which engaged participants across global hybrid locations, resulting in 34 team submissions. The submissions spanned seven key application areas and demonstrated the diverse utility of LLMs for applications in (1) molecular and material property prediction; (2) molecular and material design; (3) automation and novel interfaces; (4) scientific communication and education; (5) research data management and automation; (6) hypothesis generation and evaluation; and (7) knowledge extraction and reasoning from scientific literature. Each team submission is presented in a summary table with links to the code and as brief papers in the appendix. Beyond team results, we discuss the hackathon event and its hybrid format, which included physical hubs in Toronto, Montreal, San Francisco, Berlin, Lausanne, and Tokyo, alongside a global online hub to enable local and virtual collaboration. Overall, the event highlighted significant improvements in LLM capabilities since the previous year's hackathon, suggesting continued expansion of LLMs for applications in materials science and chemistry research. These outcomes demonstrate the dual utility of LLMs as both multipurpose models for diverse machine learning tasks and platforms for rapid prototyping custom applications in scientific research.
Establishing Deep InfoMax as an effective self-supervised learning methodology in materials informatics
Jun 30, 2024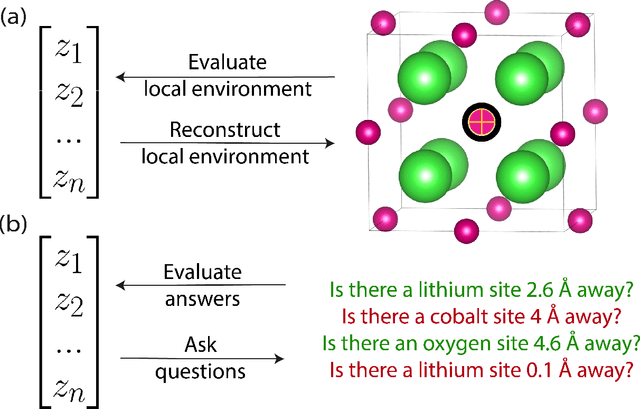

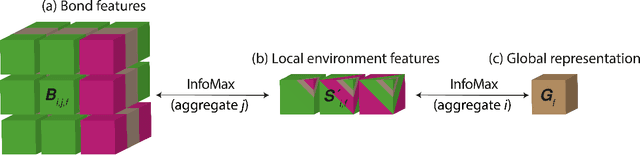

The scarcity of property labels remains a key challenge in materials informatics, whereas materials data without property labels are abundant in comparison. By pretraining supervised property prediction models on self-supervised tasks that depend only on the "intrinsic information" available in any Crystallographic Information File (CIF), there is potential to leverage the large amount of crystal data without property labels to improve property prediction results on small datasets. We apply Deep InfoMax as a self-supervised machine learning framework for materials informatics that explicitly maximises the mutual information between a point set (or graph) representation of a crystal and a vector representation suitable for downstream learning. This allows the pretraining of supervised models on large materials datasets without the need for property labels and without requiring the model to reconstruct the crystal from a representation vector. We investigate the benefits of Deep InfoMax pretraining implemented on the Site-Net architecture to improve the performance of downstream property prediction models with small amounts (<10^3) of data, a situation relevant to experimentally measured materials property databases. Using a property label masking methodology, where we perform self-supervised learning on larger supervised datasets and then train supervised models on a small subset of the labels, we isolate Deep InfoMax pretraining from the effects of distributional shift. We demonstrate performance improvements in the contexts of representation learning and transfer learning on the tasks of band gap and formation energy prediction. Having established the effectiveness of Deep InfoMax pretraining in a controlled environment, our findings provide a foundation for extending the approach to address practical challenges in materials informatics.
Element selection for functional materials discovery by integrated machine learning of atomic contributions to properties
Feb 02, 2022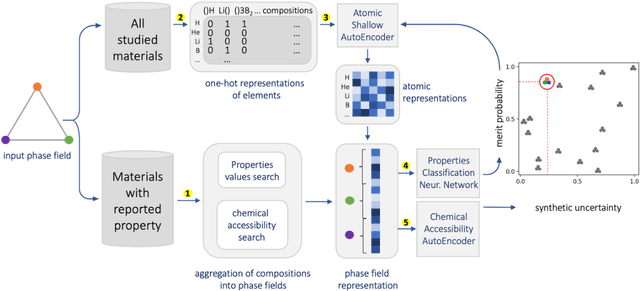
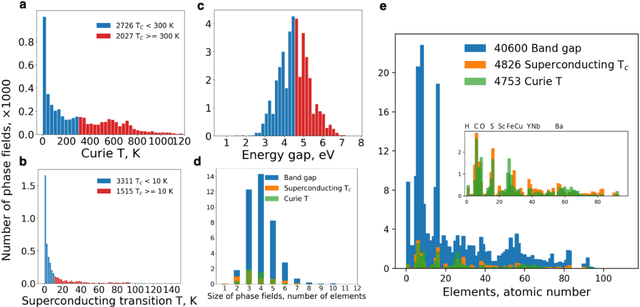
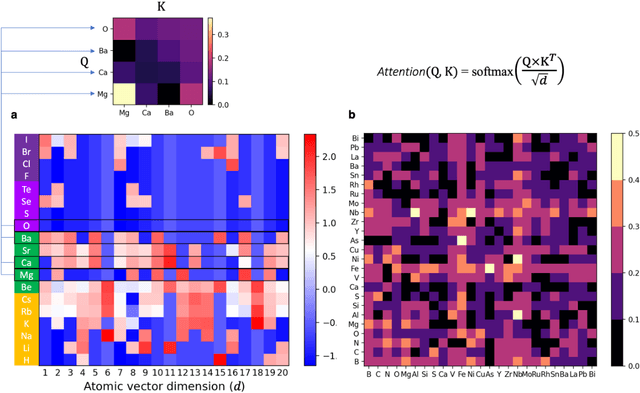
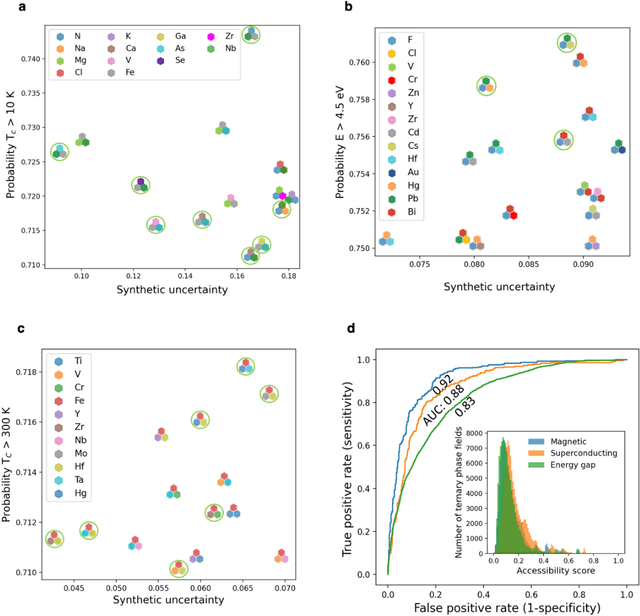
At the high level, the fundamental differences between materials originate from the unique nature of the constituent chemical elements. Before specific differences emerge according to the precise ratios of elements (composition) in a given crystal structure (phase), the material can be represented by its phase field defined simply as the set of the constituent chemical elements. Classification of the materials at the level of their phase fields can accelerate materials discovery by selecting the elemental combinations that are likely to produce desirable functional properties in synthetically accessible materials. Here, we demonstrate that classification of the materials phase field with respect to the maximum expected value of a target functional property can be combined with the ranking of the materials synthetic accessibility. This end-to-end machine learning approach (PhaseSelect) first derives the atomic characteristics from the compositional environments in all computationally and experimentally explored materials and then employs these characteristics to classify the phase field by their merit. PhaseSelect can quantify the materials potential at the level of the periodic table, which we demonstrate with significant accuracy for three avenues of materials applications: high-temperature superconducting, high-temperature magnetic and targetted energy band gap materials.