 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAssessing data-driven predictions of band gap and electrical conductivity for transparent conducting materials
Nov 21, 2024Machine Learning (ML) has offered innovative perspectives for accelerating the discovery of new functional materials, leveraging the increasing availability of material databases. Despite the promising advances, data-driven methods face constraints imposed by the quantity and quality of available data. Moreover, ML is often employed in tandem with simulated datasets originating from density functional theory (DFT), and assessed through in-sample evaluation schemes. This scenario raises questions about the practical utility of ML in uncovering new and significant material classes for industrial applications. Here, we propose a data-driven framework aimed at accelerating the discovery of new transparent conducting materials (TCMs), an important category of semiconductors with a wide range of applications. To mitigate the shortage of available data, we create and validate unique experimental databases, comprising several examples of existing TCMs. We assess state-of-the-art (SOTA) ML models for property prediction from the stoichiometry alone. We propose a bespoke evaluation scheme to provide empirical evidence on the ability of ML to uncover new, previously unseen materials of interest. We test our approach on a list of 55 compositions containing typical elements of known TCMs. Although our study indicates that ML tends to identify new TCMs compositionally similar to those in the training data, we empirically demonstrate that it can highlight material candidates that may have been previously overlooked, offering a systematic approach to identify materials that are likely to display TCMs characteristics.
Establishing Deep InfoMax as an effective self-supervised learning methodology in materials informatics
Jun 30, 2024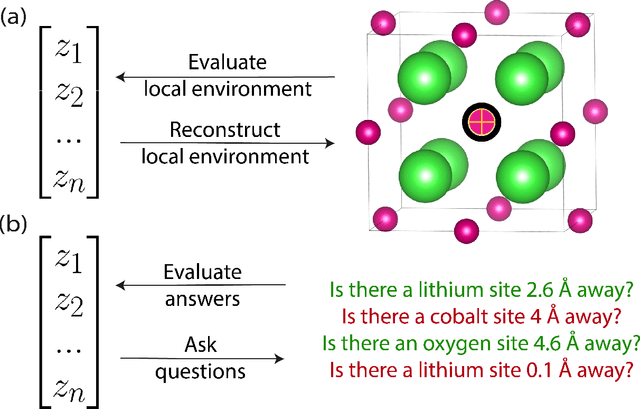

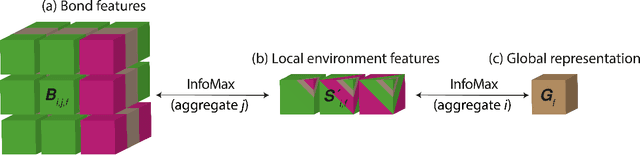

The scarcity of property labels remains a key challenge in materials informatics, whereas materials data without property labels are abundant in comparison. By pretraining supervised property prediction models on self-supervised tasks that depend only on the "intrinsic information" available in any Crystallographic Information File (CIF), there is potential to leverage the large amount of crystal data without property labels to improve property prediction results on small datasets. We apply Deep InfoMax as a self-supervised machine learning framework for materials informatics that explicitly maximises the mutual information between a point set (or graph) representation of a crystal and a vector representation suitable for downstream learning. This allows the pretraining of supervised models on large materials datasets without the need for property labels and without requiring the model to reconstruct the crystal from a representation vector. We investigate the benefits of Deep InfoMax pretraining implemented on the Site-Net architecture to improve the performance of downstream property prediction models with small amounts (<10^3) of data, a situation relevant to experimentally measured materials property databases. Using a property label masking methodology, where we perform self-supervised learning on larger supervised datasets and then train supervised models on a small subset of the labels, we isolate Deep InfoMax pretraining from the effects of distributional shift. We demonstrate performance improvements in the contexts of representation learning and transfer learning on the tasks of band gap and formation energy prediction. Having established the effectiveness of Deep InfoMax pretraining in a controlled environment, our findings provide a foundation for extending the approach to address practical challenges in materials informatics.
Metrics for quantifying isotropy in high dimensional unsupervised clustering tasks in a materials context
May 25, 2023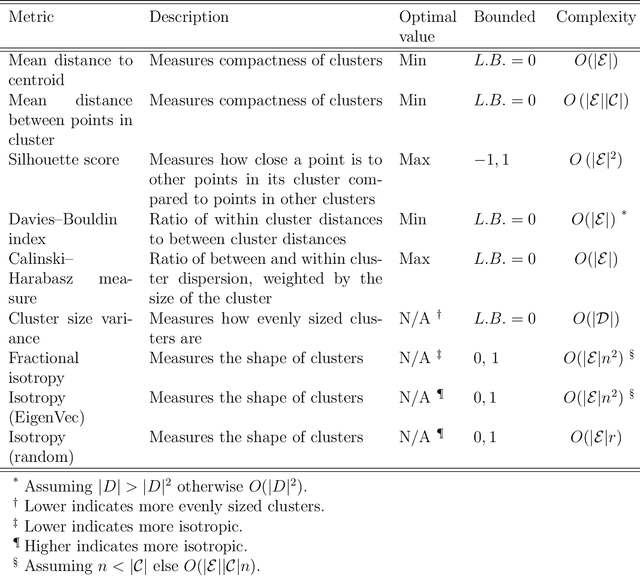
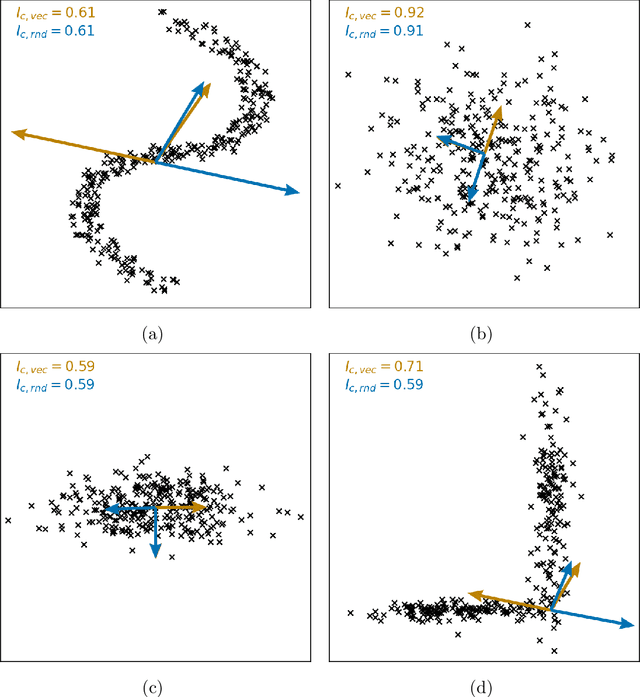
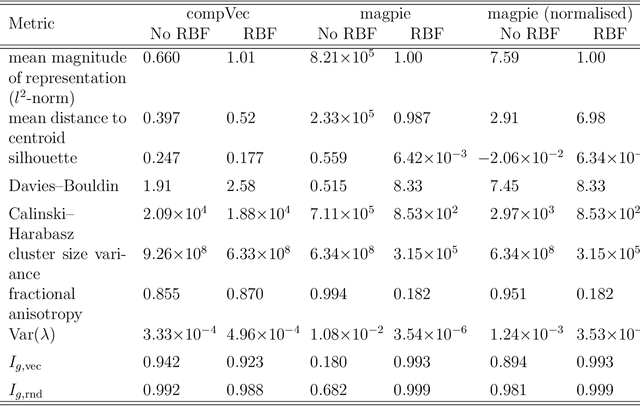
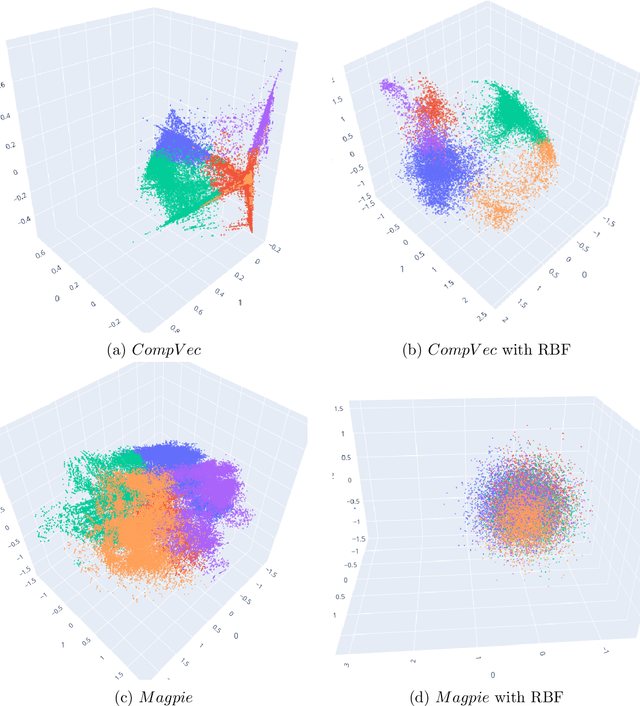
Clustering is a common task in machine learning, but clusters of unlabelled data can be hard to quantify. The application of clustering algorithms in chemistry is often dependant on material representation. Ascertaining the effects of different representations, clustering algorithms, or data transformations on the resulting clusters is difficult due to the dimensionality of these data. We present a thorough analysis of measures for isotropy of a cluster, including a novel implantation based on an existing derivation. Using fractional anisotropy, a common method used in medical imaging for comparison, we then expand these measures to examine the average isotropy of a set of clusters. A use case for such measures is demonstrated by quantifying the effects of kernel approximation functions on different representations of the Inorganic Crystal Structure Database. Broader applicability of these methods is demonstrated in analysing learnt embedding of the MNIST dataset. Random clusters are explored to examine the differences between isotropy measures presented, and to see how each method scales with the dimensionality. Python implementations of these measures are provided for use by the community.
Element selection for functional materials discovery by integrated machine learning of atomic contributions to properties
Feb 02, 2022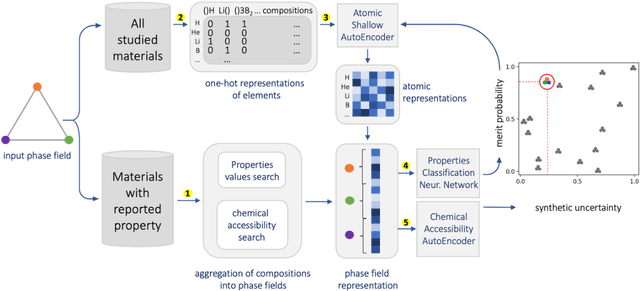
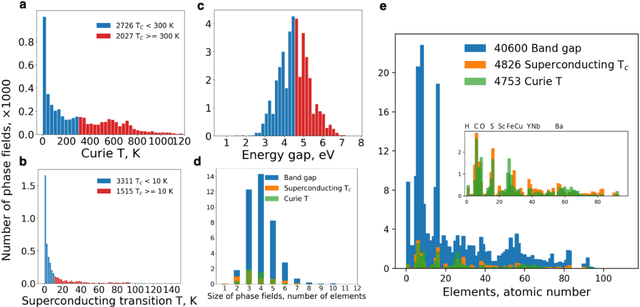
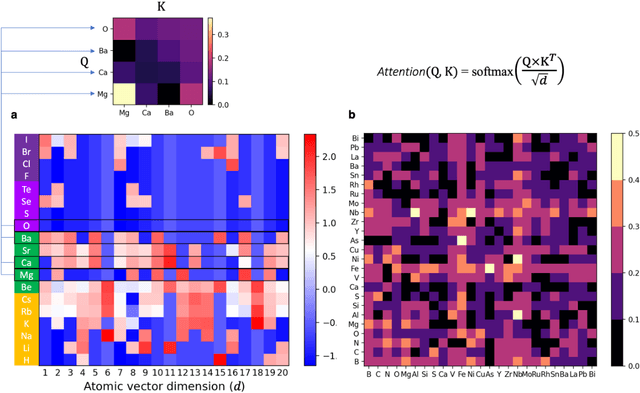
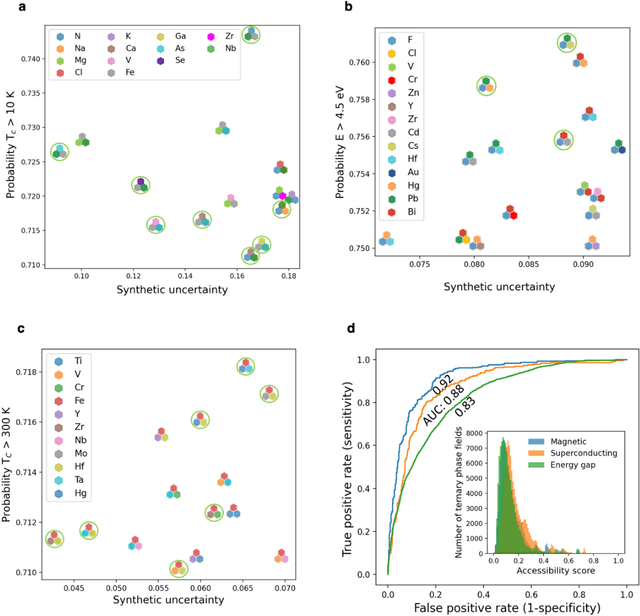
At the high level, the fundamental differences between materials originate from the unique nature of the constituent chemical elements. Before specific differences emerge according to the precise ratios of elements (composition) in a given crystal structure (phase), the material can be represented by its phase field defined simply as the set of the constituent chemical elements. Classification of the materials at the level of their phase fields can accelerate materials discovery by selecting the elemental combinations that are likely to produce desirable functional properties in synthetically accessible materials. Here, we demonstrate that classification of the materials phase field with respect to the maximum expected value of a target functional property can be combined with the ranking of the materials synthetic accessibility. This end-to-end machine learning approach (PhaseSelect) first derives the atomic characteristics from the compositional environments in all computationally and experimentally explored materials and then employs these characteristics to classify the phase field by their merit. PhaseSelect can quantify the materials potential at the level of the periodic table, which we demonstrate with significant accuracy for three avenues of materials applications: high-temperature superconducting, high-temperature magnetic and targetted energy band gap materials.