 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePredicting materials properties without crystal structure: Deep representation learning from stoichiometry
Paper and Code

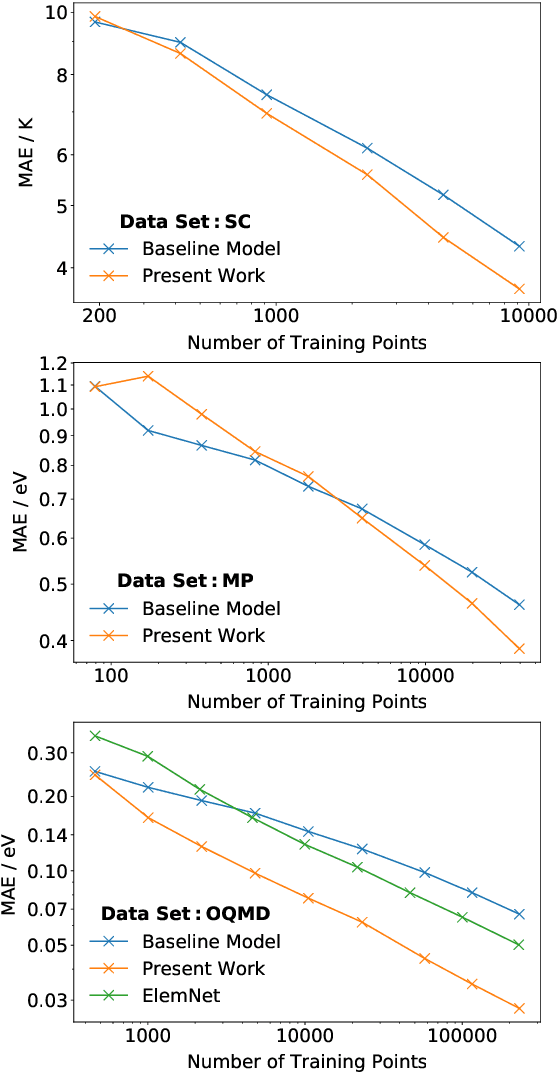
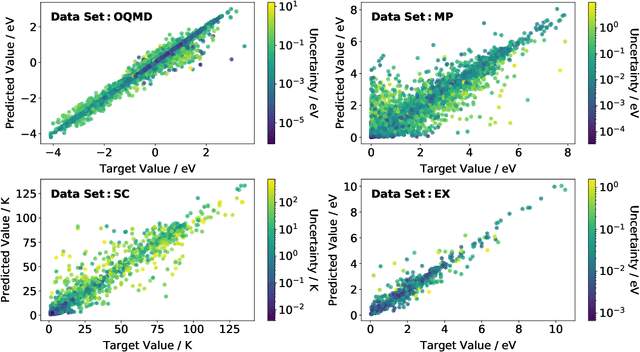
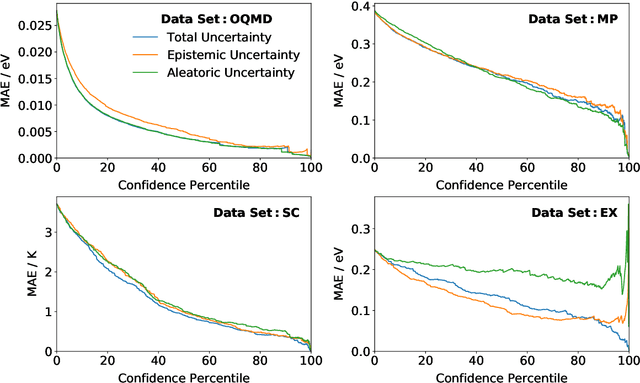
Machine learning can accelerate materials discovery by accurately predicting materials properties with low computational cost. However, the model inputs remain a key stumbling block: current methods typically use hand-engineered descriptors constructed from knowledge of either the full crystal structure -- applicable only to materials with experimentally measured structures as crystal structure prediction is computationally expensive -- or the stoichiometry. We develop a machine learning approach that takes only the stoichiometry as input and automatically learns appropriate and systematically improvable descriptors from data. Our key insight is to treat the stoichiometric formula as a dense weighted graph between elements. Compared to the state of the art, our approach achieves lower error on a plethora of challenging material properties. Moreover, our model can estimate its own uncertainty as well as transfer its learnt representation, extracting useful information from a cognate data-abundant task to deploy on a data-poor task.