 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePushing the Pareto front of band gap and permittivity: ML-guided search for dielectric materials
Jan 11, 2024



Materials with high-dielectric constant easily polarize under external electric fields, allowing them to perform essential functions in many modern electronic devices. Their practical utility is determined by two conflicting properties: high dielectric constants tend to occur in materials with narrow band gaps, limiting the operating voltage before dielectric breakdown. We present a high-throughput workflow that combines element substitution, ML pre-screening, ab initio simulation and human expert intuition to efficiently explore the vast space of unknown materials for potential dielectrics, leading to the synthesis and characterization of two novel dielectric materials, CsTaTeO6 and Bi2Zr2O7. Our key idea is to deploy ML in a multi-objective optimization setting with concave Pareto front. While usually considered more challenging than single-objective optimization, we argue and show preliminary evidence that the $1/x$-correlation between band gap and permittivity in fact makes the task more amenable to ML methods by allowing separate models for band gap and permittivity to each operate in regions of good training support while still predicting materials of exceptional merit. To our knowledge, this is the first instance of successful ML-guided multi-objective materials optimization achieving experimental synthesis and characterization. CsTaTeO6 is a structure generated via element substitution not present in our reference data sources, thus exemplifying successful de-novo materials design. Meanwhile, we report the first high-purity synthesis and dielectric characterization of Bi2Zr2O7 with a band gap of 2.27 eV and a permittivity of 20.5, meeting all target metrics of our multi-objective search.
Random projections and Kernelised Leave One Cluster Out Cross-Validation: Universal baselines and evaluation tools for supervised machine learning for materials properties
Jun 17, 2022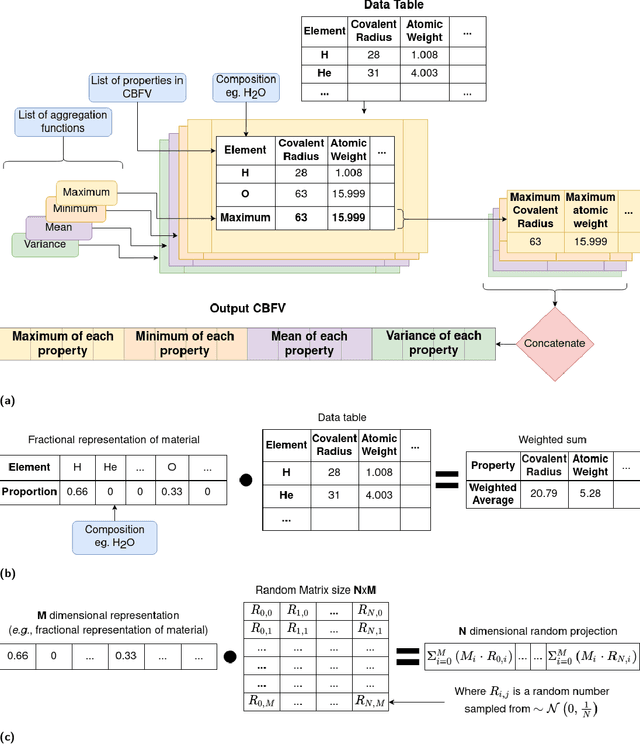
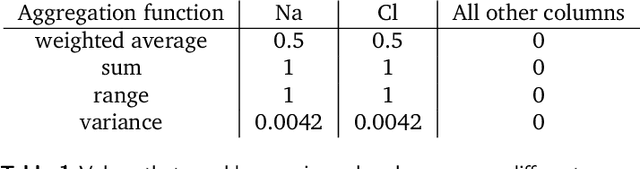
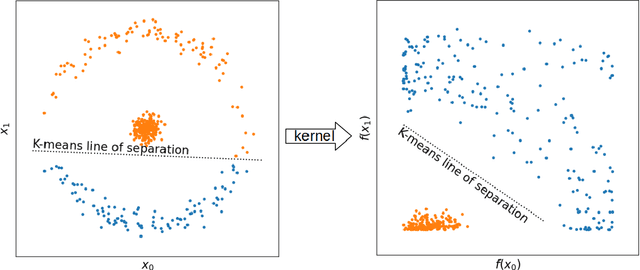
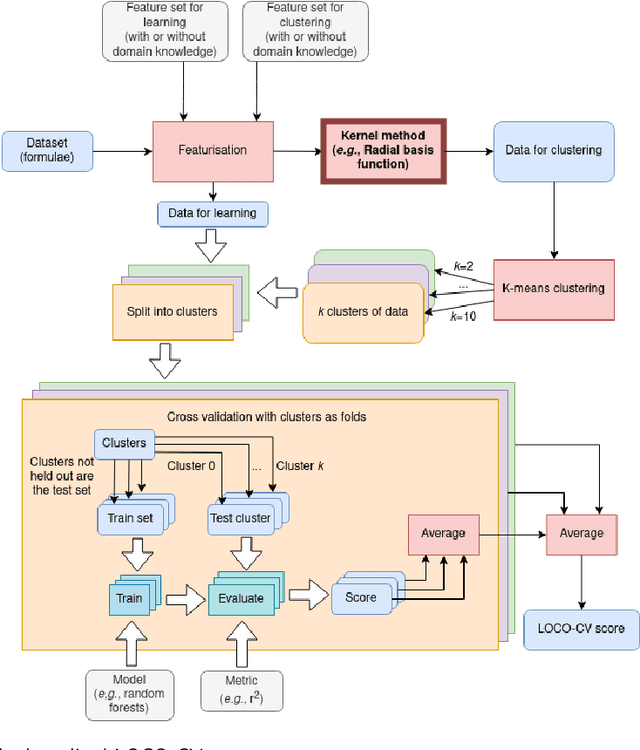
With machine learning being a popular topic in current computational materials science literature, creating representations for compounds has become common place. These representations are rarely compared, as evaluating their performance - and the performance of the algorithms that they are used with - is non-trivial. With many materials datasets containing bias and skew caused by the research process, leave one cluster out cross validation (LOCO-CV) has been introduced as a way of measuring the performance of an algorithm in predicting previously unseen groups of materials. This raises the question of the impact, and control, of the range of cluster sizes on the LOCO-CV measurement outcomes. We present a thorough comparison between composition-based representations, and investigate how kernel approximation functions can be used to better separate data to enhance LOCO-CV applications. We find that domain knowledge does not improve machine learning performance in most tasks tested, with band gap prediction being the notable exception. We also find that the radial basis function improves the linear separability of chemical datasets in all 10 datasets tested and provide a framework for the application of this function in the LOCO-CV process to improve the outcome of LOCO-CV measurements regardless of machine learning algorithm, choice of metric, and choice of compound representation. We recommend kernelised LOCO-CV as a training paradigm for those looking to measure the extrapolatory power of an algorithm on materials data.