 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePushing the Pareto front of band gap and permittivity: ML-guided search for dielectric materials
Jan 11, 2024Materials with high-dielectric constant easily polarize under external electric fields, allowing them to perform essential functions in many modern electronic devices. Their practical utility is determined by two conflicting properties: high dielectric constants tend to occur in materials with narrow band gaps, limiting the operating voltage before dielectric breakdown. We present a high-throughput workflow that combines element substitution, ML pre-screening, ab initio simulation and human expert intuition to efficiently explore the vast space of unknown materials for potential dielectrics, leading to the synthesis and characterization of two novel dielectric materials, CsTaTeO6 and Bi2Zr2O7. Our key idea is to deploy ML in a multi-objective optimization setting with concave Pareto front. While usually considered more challenging than single-objective optimization, we argue and show preliminary evidence that the $1/x$-correlation between band gap and permittivity in fact makes the task more amenable to ML methods by allowing separate models for band gap and permittivity to each operate in regions of good training support while still predicting materials of exceptional merit. To our knowledge, this is the first instance of successful ML-guided multi-objective materials optimization achieving experimental synthesis and characterization. CsTaTeO6 is a structure generated via element substitution not present in our reference data sources, thus exemplifying successful de-novo materials design. Meanwhile, we report the first high-purity synthesis and dielectric characterization of Bi2Zr2O7 with a band gap of 2.27 eV and a permittivity of 20.5, meeting all target metrics of our multi-objective search.
Molecular Transformer for Chemical Reaction Prediction and Uncertainty Estimation
Nov 06, 2018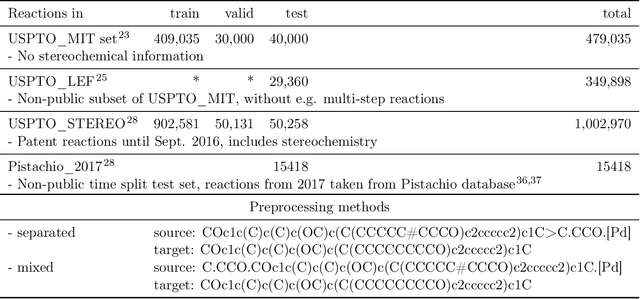
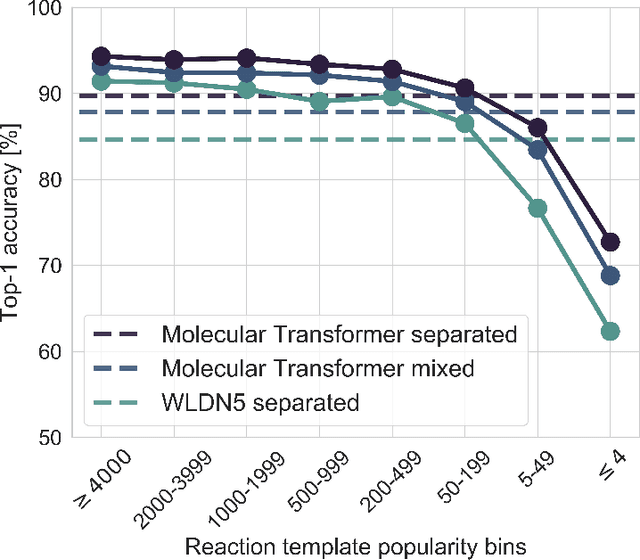
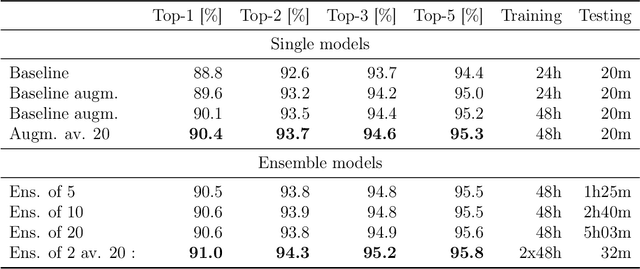
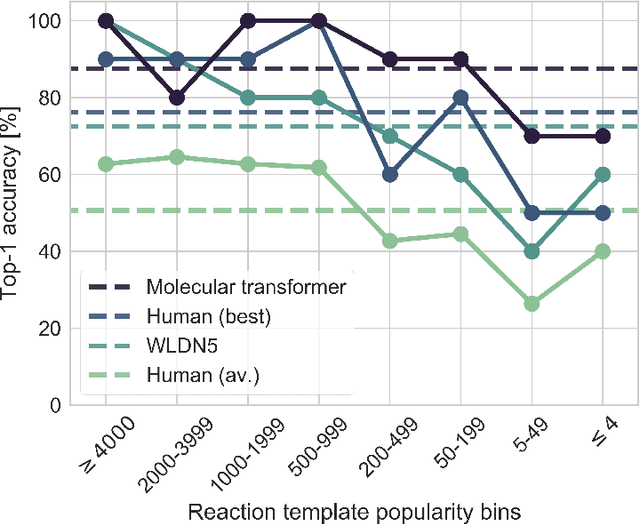
Organic synthesis is one of the key stumbling blocks in medicinal chemistry. A necessary yet unsolved step in planning synthesis is solving the forward problem: given reactants and reagents, predict the products. Similar to other works, we treat reaction prediction as a machine translation problem between SMILES strings of reactants-reagents and the products. We show that a multi-head attention Molecular Transformer model outperforms all algorithms in the literature, achieving a top-1 accuracy above 90% on a common benchmark dataset. Our algorithm requires no handcrafted rules, and accurately predicts subtle chemical transformations. Crucially, our model can accurately estimate its own uncertainty, with an uncertainty score that is 89% accurate in terms of classifying whether a prediction is correct. Furthermore, we show that the model is able to handle inputs without reactant-reagent split and including stereochemistry, which makes our method universally applicable.