 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFrom Knowledge to Action: Outcomes of the 2025 Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry
May 04, 2026Large language models (LLMs) are rapidly changing how researchers in materials science and chemistry discover, organize, and act on scientific knowledge. This paper analyzes a broad set of community-developed LLM applications in an effort to identify emerging patterns in how these systems can be used across the scientific research lifecycle. We organize the projects into two complementary categories: Knowledge Infrastructure, systems that structure, retrieve, synthesize, and validate scientific information; and Action Systems, systems that execute, coordinate, or automate scientific work across computational and experimental environments. The submissions reveal a shift from single-purpose LLM tools toward integrated, multi-agent workflows that combine retrieval, reasoning, tool use, and domain-specific validation. Prominent themes include retrieval-augmented generation as grounding infrastructure, persistent structured knowledge representations, multimodal and multilingual scientific inputs, and early progress toward laboratory-integrated closed-loop systems. Together, these results suggest that LLMs are evolving from general-purpose assistants into composable infrastructure for scientific reasoning and action. This work provides a community snapshot of that transition and a practical taxonomy for understanding emerging LLM-enabled workflows in materials science and chemistry.
CASCADE: Cumulative Agentic Skill Creation through Autonomous Development and Evolution
Dec 29, 2025Large language model (LLM) agents currently depend on predefined tools or brittle tool generation, constraining their capability and adaptability to complex scientific tasks. We introduce CASCADE, a self-evolving agentic framework representing an early instantiation of the transition from "LLM + tool use" to "LLM + skill acquisition". CASCADE enables agents to master complex external tools and codify knowledge through two meta-skills: continuous learning via web search and code extraction, and self-reflection via introspection and knowledge graph exploration, among others. We evaluate CASCADE on SciSkillBench, a benchmark of 116 materials science and chemistry research tasks. CASCADE achieves a 93.3% success rate using GPT-5, compared to 35.4% without evolution mechanisms. We further demonstrate real-world applications in computational analysis, autonomous laboratory experiments, and selective reproduction of published papers. Along with human-agent collaboration and memory consolidation, CASCADE accumulates executable skills that can be shared across agents and scientists, moving toward scalable AI-assisted scientific research.
MiST: Understanding the Role of Mid-Stage Scientific Training in Developing Chemical Reasoning Models
Dec 24, 2025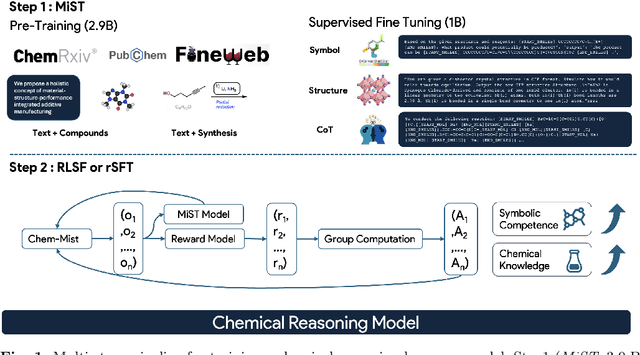
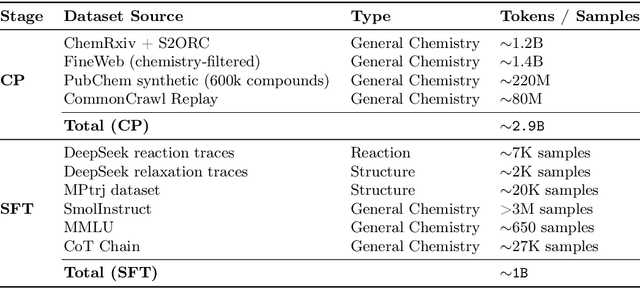
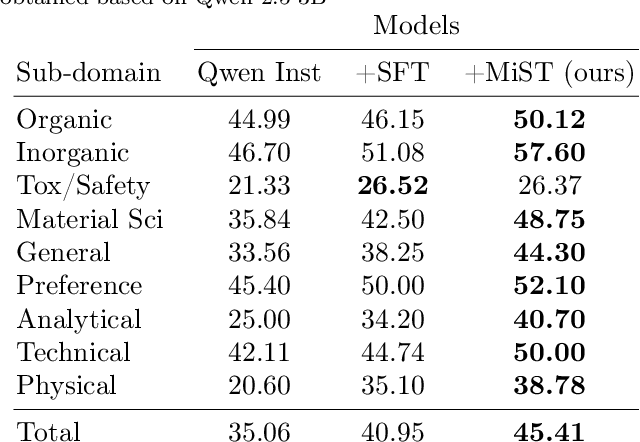
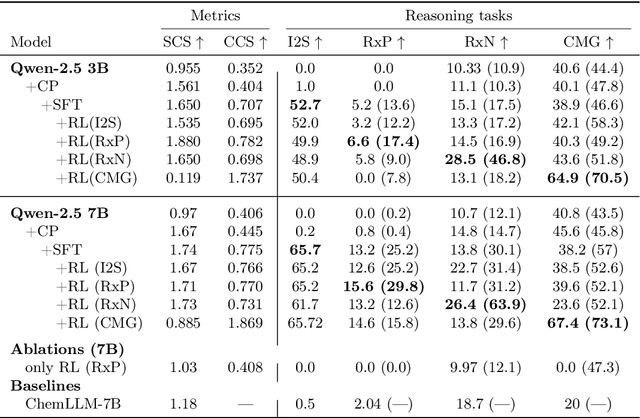
Large Language Models can develop reasoning capabilities through online fine-tuning with rule-based rewards. However, recent studies reveal a critical constraint: reinforcement learning succeeds only when the base model already assigns non-negligible probability to correct answers -- a property we term 'latent solvability'. This work investigates the emergence of chemical reasoning capabilities and what these prerequisites mean for chemistry. We identify two necessary conditions for RL-based chemical reasoning: 1) Symbolic competence, and 2) Latent chemical knowledge. We propose mid-stage scientific training (MiST): a set of mid-stage training techniques to satisfy these, including data-mixing with SMILES/CIF-aware pre-processing, continued pre-training on 2.9B tokens, and supervised fine-tuning on 1B tokens. These steps raise the latent-solvability score on 3B and 7B models by up to 1.8x, and enable RL to lift top-1 accuracy from 10.9 to 63.9% on organic reaction naming, and from 40.6 to 67.4% on inorganic material generation. Similar results are observed for other challenging chemical tasks, while producing interpretable reasoning traces. Our results define clear prerequisites for chemical reasoning training and highlight the broader role of mid-stage training in unlocking reasoning capabilities.
Synthelite: Chemist-aligned and feasibility-aware synthesis planning with LLMs
Dec 18, 2025



Computer-aided synthesis planning (CASP) has long been envisioned as a complementary tool for synthetic chemists. However, existing frameworks often lack mechanisms to allow interaction with human experts, limiting their ability to integrate chemists' insights. In this work, we introduce Synthelite, a synthesis planning framework that uses large language models (LLMs) to directly propose retrosynthetic transformations. Synthelite can generate end-to-end synthesis routes by harnessing the intrinsic chemical knowledge and reasoning capabilities of LLMs, while allowing expert intervention through natural language prompts. Our experiments demonstrate that Synthelite can flexibly adapt its planning trajectory to diverse user-specified constraints, achieving up to 95\% success rates in both strategy-constrained and starting-material-constrained synthesis tasks. Additionally, Synthelite exhibits the ability to account for chemical feasibility during route design. We envision Synthelite to be both a useful tool and a step toward a paradigm where LLMs are the central orchestrators of synthesis planning.
DynaMate: An Autonomous Agent for Protein-Ligand Molecular Dynamics Simulations
Dec 10, 2025Force field-based molecular dynamics (MD) simulations are indispensable for probing the structure, dynamics, and functions of biomolecular systems, including proteins and protein-ligand complexes. Despite their broad utility in drug discovery and protein engineering, the technical complexity of MD setup, encompassing parameterization, input preparation, and software configuration, remains a major barrier for widespread and efficient usage. Agentic LLMs have demonstrated their capacity to autonomously execute multi-step scientific processes, and to date, they have not successfully been used to automate protein-ligand MD workflows. Here, we present DynaMate, a modular multi-agent framework that autonomously designs and executes complete MD workflows for both protein and protein-ligand systems, and offers free energy binding affinity calculations with the MM/PB(GB)SA method. The framework integrates dynamic tool use, web search, PaperQA, and a self-correcting behavior. DynaMate comprises three specialized modules, interacting to plan the experiment, perform the simulation, and analyze the results. We evaluated its performance across twelve benchmark systems of varying complexity, assessing success rate, efficiency, and adaptability. DynaMate reliably performed full MD simulations, corrected runtime errors through iterative reasoning, and produced meaningful analyses of protein-ligand interactions. This automated framework paves the way toward standardized, scalable, and time-efficient molecular modeling pipelines for future biomolecular and drug design applications.
Flow-Based Fragment Identification via Binding Site-Specific Latent Representations
Sep 16, 2025Fragment-based drug design is a promising strategy leveraging the binding of small chemical moieties that can efficiently guide drug discovery. The initial step of fragment identification remains challenging, as fragments often bind weakly and non-specifically. We developed a protein-fragment encoder that relies on a contrastive learning approach to map both molecular fragments and protein surfaces in a shared latent space. The encoder captures interaction-relevant features and allows to perform virtual screening as well as generative design with our new method LatentFrag. In LatentFrag, fragment embeddings and positions are generated conditioned on the protein surface while being chemically realistic by construction. Our expressive fragment and protein representations allow location of protein-fragment interaction sites with high sensitivity and we observe state-of-the-art fragment recovery rates when sampling from the learned distribution of latent fragment embeddings. Our generative method outperforms common methods such as virtual screening at a fraction of its computational cost providing a valuable starting point for fragment hit discovery. We further show the practical utility of LatentFrag and extend the workflow to full ligand design tasks. Together, these approaches contribute to advancing fragment identification and provide valuable tools for fragment-based drug discovery.
Lookup multivariate Kolmogorov-Arnold Networks
Sep 08, 2025



High-dimensional linear mappings, or linear layers, dominate both the parameter count and the computational cost of most modern deep-learning models. We introduce a general drop-in replacement, lookup multivariate Kolmogorov-Arnold Networks (lmKANs), which deliver a substantially better trade-off between capacity and inference cost. Our construction expresses a general high-dimensional mapping through trainable low-dimensional multivariate functions. These functions can carry dozens or hundreds of trainable parameters each, and yet it takes only a few multiplications to compute them because they are implemented as spline lookup tables. Empirically, lmKANs reduce inference FLOPs by up to 6.0x while matching the flexibility of MLPs in general high-dimensional function approximation. In another feedforward fully connected benchmark, on the tabular-like dataset of randomly displaced methane configurations, lmKANs enable more than 10x higher H100 throughput at equal accuracy. Within frameworks of Convolutional Neural Networks, lmKAN-based CNNs cut inference FLOPs at matched accuracy by 1.6-2.1x and by 1.7x on the CIFAR-10 and ImageNet-1k datasets, respectively. Our code, including dedicated CUDA kernels, is available online at https://github.com/schwallergroup/lmkan.
Position: Intelligent Science Laboratory Requires the Integration of Cognitive and Embodied AI
Jun 24, 2025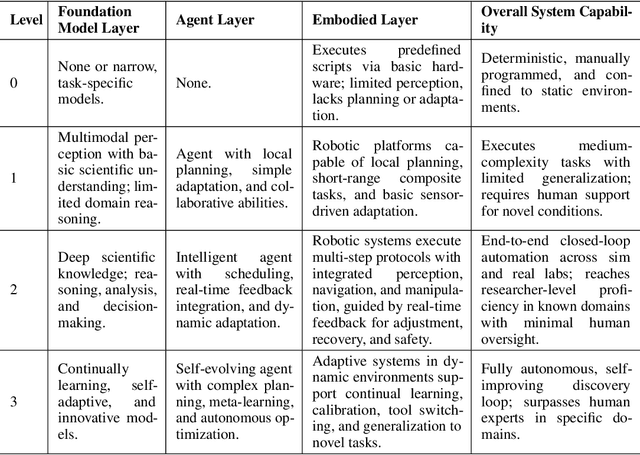
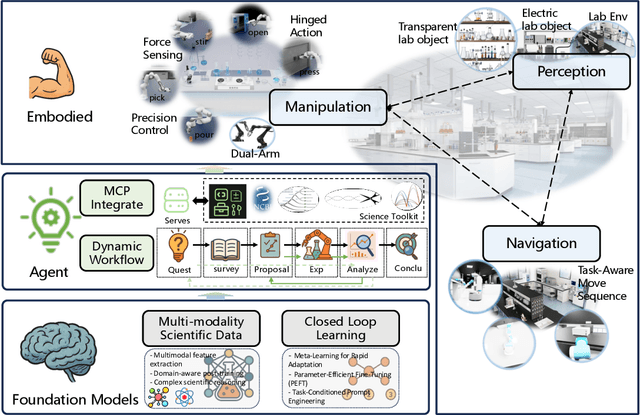
Scientific discovery has long been constrained by human limitations in expertise, physical capability, and sleep cycles. The recent rise of AI scientists and automated laboratories has accelerated both the cognitive and operational aspects of research. However, key limitations persist: AI systems are often confined to virtual environments, while automated laboratories lack the flexibility and autonomy to adaptively test new hypotheses in the physical world. Recent advances in embodied AI, such as generalist robot foundation models, diffusion-based action policies, fine-grained manipulation learning, and sim-to-real transfer, highlight the promise of integrating cognitive and embodied intelligence. This convergence opens the door to closed-loop systems that support iterative, autonomous experimentation and the possibility of serendipitous discovery. In this position paper, we propose the paradigm of Intelligent Science Laboratories (ISLs): a multi-layered, closed-loop framework that deeply integrates cognitive and embodied intelligence. ISLs unify foundation models for scientific reasoning, agent-based workflow orchestration, and embodied agents for robust physical experimentation. We argue that such systems are essential for overcoming the current limitations of scientific discovery and for realizing the full transformative potential of AI-driven science.
ChemPile: A 250GB Diverse and Curated Dataset for Chemical Foundation Models
May 18, 2025



Foundation models have shown remarkable success across scientific domains, yet their impact in chemistry remains limited due to the absence of diverse, large-scale, high-quality datasets that reflect the field's multifaceted nature. We present the ChemPile, an open dataset containing over 75 billion tokens of curated chemical data, specifically built for training and evaluating general-purpose models in the chemical sciences. The dataset mirrors the human learning journey through chemistry -- from educational foundations to specialized expertise -- spanning multiple modalities and content types including structured data in diverse chemical representations (SMILES, SELFIES, IUPAC names, InChI, molecular renderings), scientific and educational text, executable code, and chemical images. ChemPile integrates foundational knowledge (textbooks, lecture notes), specialized expertise (scientific articles and language-interfaced data), visual understanding (molecular structures, diagrams), and advanced reasoning (problem-solving traces and code) -- mirroring how human chemists develop expertise through diverse learning materials and experiences. Constructed through hundreds of hours of expert curation, the ChemPile captures both foundational concepts and domain-specific complexity. We provide standardized training, validation, and test splits, enabling robust benchmarking. ChemPile is openly released via HuggingFace with a consistent API, permissive license, and detailed documentation. We hope the ChemPile will serve as a catalyst for chemical AI, enabling the development of the next generation of chemical foundation models.
Generative Molecular Design with Steerable and Granular Synthesizability Control
May 13, 2025Synthesizability in small molecule generative design remains a bottleneck. Existing works that do consider synthesizability can output predicted synthesis routes for generated molecules. However, there has been minimal attention in addressing the ease of synthesis and enabling flexibility to incorporate desired reaction constraints. In this work, we propose a small molecule generative design framework that enables steerable and granular synthesizability control. Generated molecules satisfy arbitrary multi-parameter optimization objectives with predicted synthesis routes containing pre-defined allowed reactions, while optionally avoiding others. One can also enforce that all reactions belong to a pre-defined set. We show the capability to mix-and-match these reaction constraints across the most common medicinal chemistry transformations. Next, we show how our framework can be used to valorize industrial byproducts towards de novo optimized molecules. Going further, we demonstrate how granular control over synthesizability constraints can loosely mimic virtual screening of ultra-large make-on-demand libraries. Using only a single GPU, we generate and dock 15k molecules to identify promising candidates in Freedom 4.0 constituting 142B make-on-demand molecules (assessing only 0.00001% of the library). Generated molecules satisfying the reaction constraints have > 90% exact match rate. Lastly, we benchmark our framework against recent synthesizability-constrained generative models and demonstrate the highest sample efficiency even when imposing the additional constraint that all molecules must be synthesizable from a single reaction type. The main theme is demonstrating that a pre-trained generalist molecular generative model can be incentivized to generate property-optimized small molecules under challenging synthesizability constraints through reinforcement learning.