 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGenerative Molecular Design with Steerable and Granular Synthesizability Control
May 13, 2025Synthesizability in small molecule generative design remains a bottleneck. Existing works that do consider synthesizability can output predicted synthesis routes for generated molecules. However, there has been minimal attention in addressing the ease of synthesis and enabling flexibility to incorporate desired reaction constraints. In this work, we propose a small molecule generative design framework that enables steerable and granular synthesizability control. Generated molecules satisfy arbitrary multi-parameter optimization objectives with predicted synthesis routes containing pre-defined allowed reactions, while optionally avoiding others. One can also enforce that all reactions belong to a pre-defined set. We show the capability to mix-and-match these reaction constraints across the most common medicinal chemistry transformations. Next, we show how our framework can be used to valorize industrial byproducts towards de novo optimized molecules. Going further, we demonstrate how granular control over synthesizability constraints can loosely mimic virtual screening of ultra-large make-on-demand libraries. Using only a single GPU, we generate and dock 15k molecules to identify promising candidates in Freedom 4.0 constituting 142B make-on-demand molecules (assessing only 0.00001% of the library). Generated molecules satisfying the reaction constraints have > 90% exact match rate. Lastly, we benchmark our framework against recent synthesizability-constrained generative models and demonstrate the highest sample efficiency even when imposing the additional constraint that all molecules must be synthesizable from a single reaction type. The main theme is demonstrating that a pre-trained generalist molecular generative model can be incentivized to generate property-optimized small molecules under challenging synthesizability constraints through reinforcement learning.
Best Practices for Multi-Fidelity Bayesian Optimization in Materials and Molecular Research
Oct 01, 2024



Multi-fidelity Bayesian Optimization (MFBO) is a promising framework to speed up materials and molecular discovery as sources of information of different accuracies are at hand at increasing cost. Despite its potential use in chemical tasks, there is a lack of systematic evaluation of the many parameters playing a role in MFBO. In this work, we provide guidelines and recommendations to decide when to use MFBO in experimental settings. We investigate MFBO methods applied to molecules and materials problems. First, we test two different families of acquisition functions in two synthetic problems and study the effect of the informativeness and cost of the approximate function. We use our implementation and guidelines to benchmark three real discovery problems and compare them against their single-fidelity counterparts. Our results may help guide future efforts to implement MFBO as a routine tool in the chemical sciences.
Holistic chemical evaluation reveals pitfalls in reaction prediction models
Dec 14, 2023
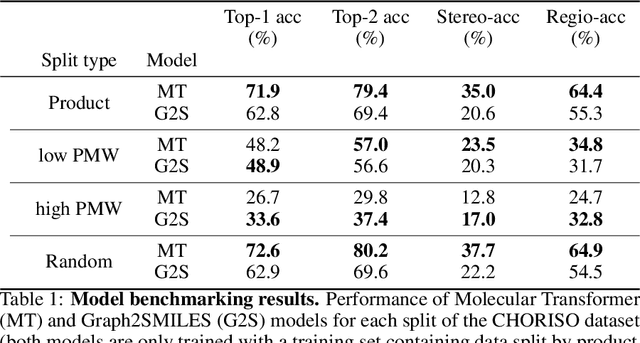


The prediction of chemical reactions has gained significant interest within the machine learning community in recent years, owing to its complexity and crucial applications in chemistry. However, model evaluation for this task has been mostly limited to simple metrics like top-k accuracy, which obfuscates fine details of a model's limitations. Inspired by progress in other fields, we propose a new assessment scheme that builds on top of current approaches, steering towards a more holistic evaluation. We introduce the following key components for this goal: CHORISO, a curated dataset along with multiple tailored splits to recreate chemically relevant scenarios, and a collection of metrics that provide a holistic view of a model's advantages and limitations. Application of this method to state-of-the-art models reveals important differences on sensitive fronts, especially stereoselectivity and chemical out-of-distribution generalization. Our work paves the way towards robust prediction models that can ultimately accelerate chemical discovery.