 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTransforming the Hybrid Cloud for Emerging AI Workloads
Nov 20, 2024



This white paper, developed through close collaboration between IBM Research and UIUC researchers within the IIDAI Institute, envisions transforming hybrid cloud systems to meet the growing complexity of AI workloads through innovative, full-stack co-design approaches, emphasizing usability, manageability, affordability, adaptability, efficiency, and scalability. By integrating cutting-edge technologies such as generative and agentic AI, cross-layer automation and optimization, unified control plane, and composable and adaptive system architecture, the proposed framework addresses critical challenges in energy efficiency, performance, and cost-effectiveness. Incorporating quantum computing as it matures will enable quantum-accelerated simulations for materials science, climate modeling, and other high-impact domains. Collaborative efforts between academia and industry are central to this vision, driving advancements in foundation models for material design and climate solutions, scalable multimodal data processing, and enhanced physics-based AI emulators for applications like weather forecasting and carbon sequestration. Research priorities include advancing AI agentic systems, LLM as an Abstraction (LLMaaA), AI model optimization and unified abstractions across heterogeneous infrastructure, end-to-end edge-cloud transformation, efficient programming model, middleware and platform, secure infrastructure, application-adaptive cloud systems, and new quantum-classical collaborative workflows. These ideas and solutions encompass both theoretical and practical research questions, requiring coordinated input and support from the research community. This joint initiative aims to establish hybrid clouds as secure, efficient, and sustainable platforms, fostering breakthroughs in AI-driven applications and scientific discovery across academia, industry, and society.
Language models in molecular discovery
Sep 28, 2023The success of language models, especially transformer-based architectures, has trickled into other domains giving rise to "scientific language models" that operate on small molecules, proteins or polymers. In chemistry, language models contribute to accelerating the molecule discovery cycle as evidenced by promising recent findings in early-stage drug discovery. Here, we review the role of language models in molecular discovery, underlining their strength in de novo drug design, property prediction and reaction chemistry. We highlight valuable open-source software assets thus lowering the entry barrier to the field of scientific language modeling. Last, we sketch a vision for future molecular design that combines a chatbot interface with access to computational chemistry tools. Our contribution serves as a valuable resource for researchers, chemists, and AI enthusiasts interested in understanding how language models can and will be used to accelerate chemical discovery.
Unifying Molecular and Textual Representations via Multi-task Language Modelling
Jan 29, 2023The recent advances in neural language models have also been successfully applied to the field of chemistry, offering generative solutions for classical problems in molecular design and synthesis planning. These new methods have the potential to optimize laboratory operations and fuel a new era of data-driven automation in scientific discovery. However, specialized models are still typically required for each task, leading to the need for problem-specific fine-tuning and neglecting task interrelations. The main obstacle in this field is the lack of a unified representation between natural language and chemical representations, complicating and limiting human-machine interaction. Here, we propose a multi-domain, multi-task language model to solve a wide range of tasks in both the chemical and natural language domains. By leveraging multi-task learning, our model can handle chemical and natural language concurrently, without requiring expensive pre-training on single domains or task-specific models. Interestingly, sharing weights across domains remarkably improves our model when benchmarked against state-of-the-art baselines on single-domain and cross-domain tasks. In particular, sharing information across domains and tasks gives rise to large improvements in cross-domain tasks, the magnitude of which increase with scale, as measured by more than a dozen of relevant metrics. Our work suggests that such models can robustly and efficiently accelerate discovery in physical sciences by superseding problem-specific fine-tuning and enhancing human-model interactions.
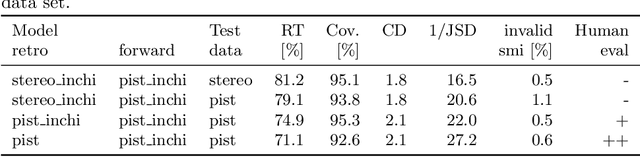
Unassisted Noise Reduction of Chemical Reaction Data Sets
Feb 02, 2021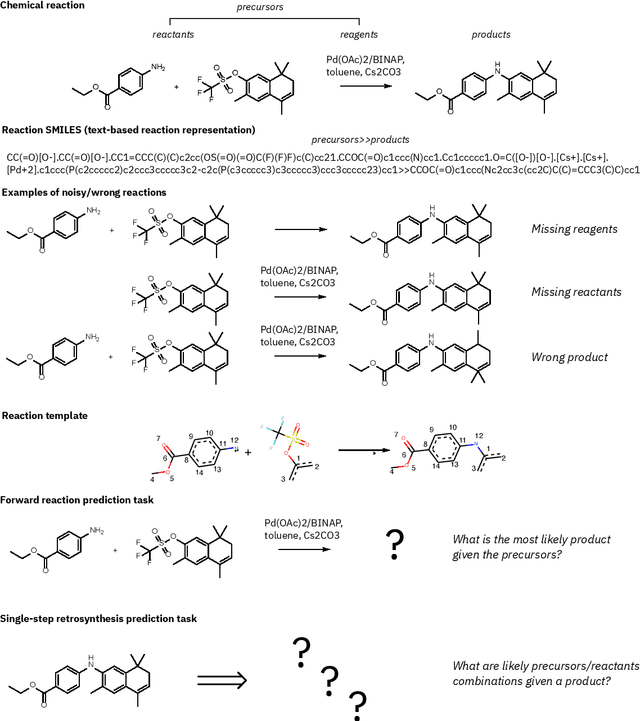
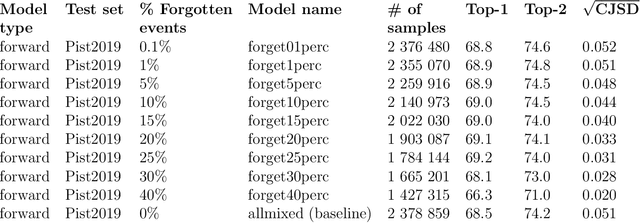
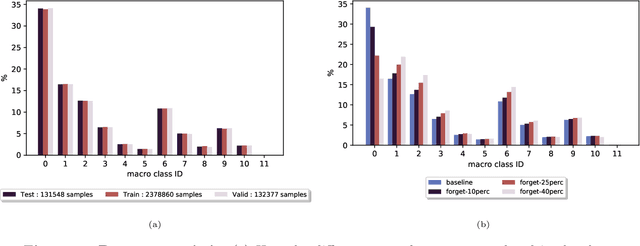

Existing deep learning models applied to reaction prediction in organic chemistry can reach high levels of accuracy (> 90% for Natural Language Processing-based ones). With no chemical knowledge embedded than the information learnt from reaction data, the quality of the data sets plays a crucial role in the performance of the prediction models. While human curation is prohibitively expensive, the need for unaided approaches to remove chemically incorrect entries from existing data sets is essential to improve artificial intelligence models' performance in synthetic chemistry tasks. Here we propose a machine learning-based, unassisted approach to remove chemically wrong entries from chemical reaction collections. We applied this method to the collection of chemical reactions Pistachio and to an open data set, both extracted from USPTO (United States Patent Office) patents. Our results show an improved prediction quality for models trained on the cleaned and balanced data sets. For the retrosynthetic models, the round-trip accuracy metric grows by 13 percentage points and the value of the cumulative Jensen Shannon divergence decreases by 30% compared to its original record. The coverage remains high with 97%, and the value of the class-diversity is not affected by the cleaning. The proposed strategy is the first unassisted rule-free technique to address automatic noise reduction in chemical data sets.
Mapping the Space of Chemical Reactions Using Attention-Based Neural Networks
Dec 09, 2020
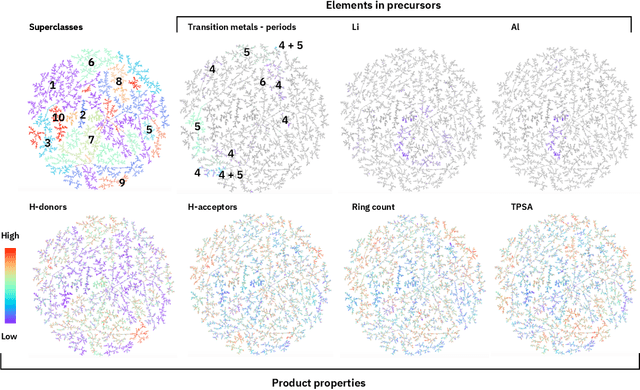
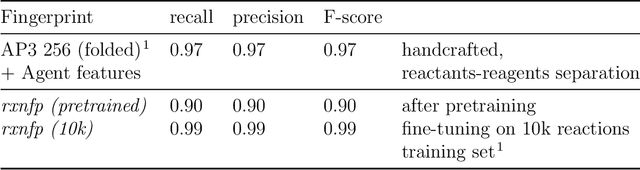

Organic reactions are usually assigned to classes containing reactions with similar reagents and mechanisms. Reaction classes facilitate the communication of complex concepts and efficient navigation through chemical reaction space. However, the classification process is a tedious task. It requires the identification of the corresponding reaction class template via annotation of the number of molecules in the reactions, the reaction center, and the distinction between reactants and reagents. This work shows that transformer-based models can infer reaction classes from non-annotated, simple text-based representations of chemical reactions. Our best model reaches a classification accuracy of 98.2%. We also show that the learned representations can be used as reaction fingerprints that capture fine-grained differences between reaction classes better than traditional reaction fingerprints. The insights into chemical reaction space enabled by our learned fingerprints are illustrated by an interactive reaction atlas providing visual clustering and similarity searching.
Exploring Chemical Space using Natural Language Processing Methodologies for Drug Discovery
Feb 10, 2020
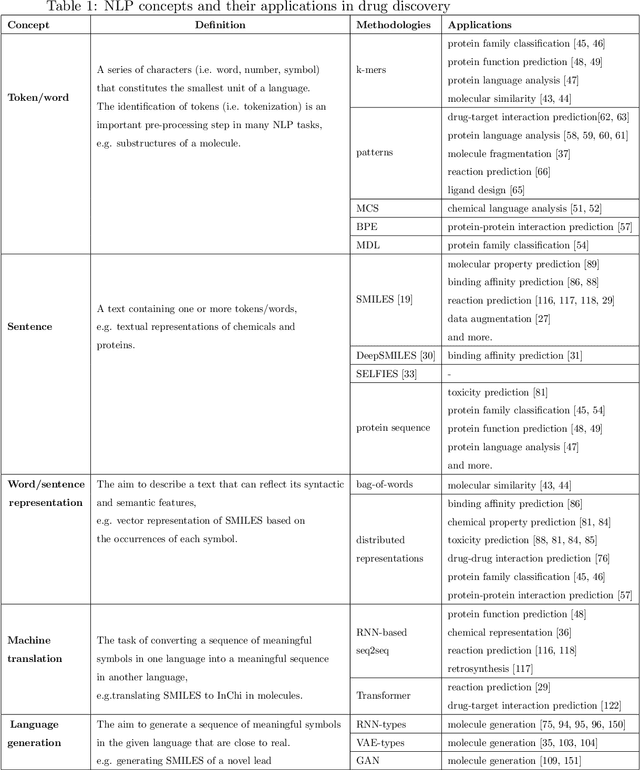
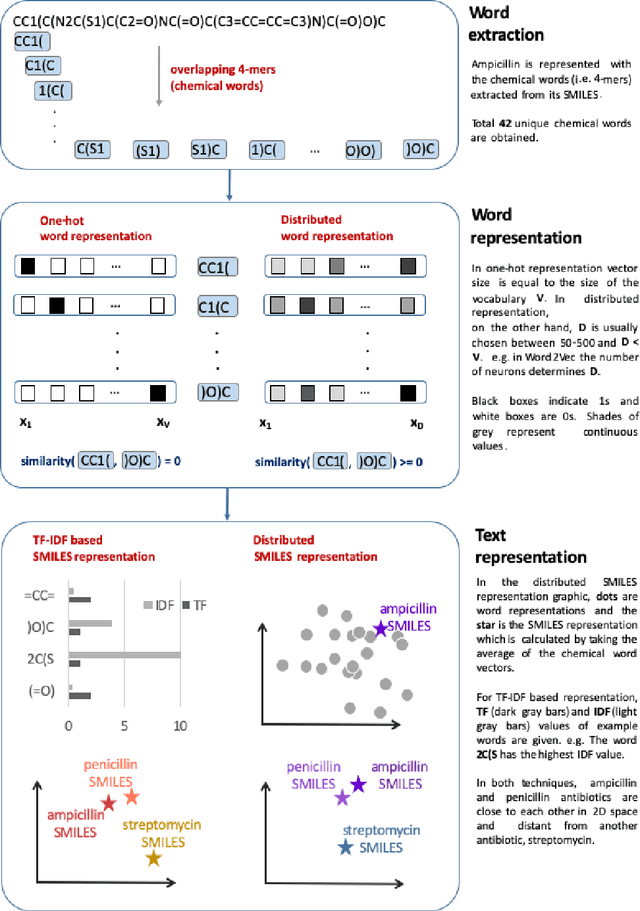
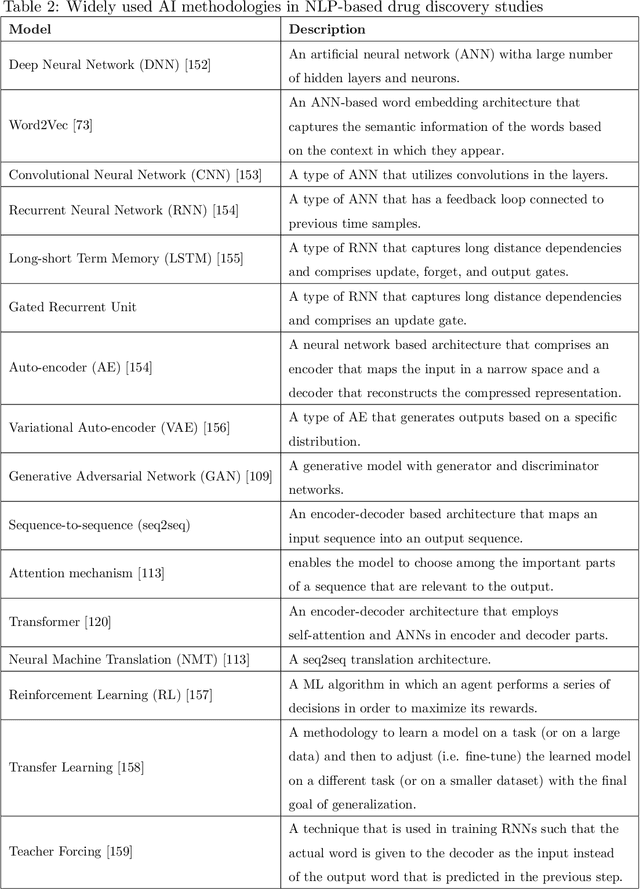
Text-based representations of chemicals and proteins can be thought of as unstructured languages codified by humans to describe domain-specific knowledge. Advances in natural language processing (NLP) methodologies in the processing of spoken languages accelerated the application of NLP to elucidate hidden knowledge in textual representations of these biochemical entities and then use it to construct models to predict molecular properties or to design novel molecules. This review outlines the impact made by these advances on drug discovery and aims to further the dialogue between medicinal chemists and computer scientists.
Predicting retrosynthetic pathways using a combined linguistic model and hyper-graph exploration strategy
Oct 17, 2019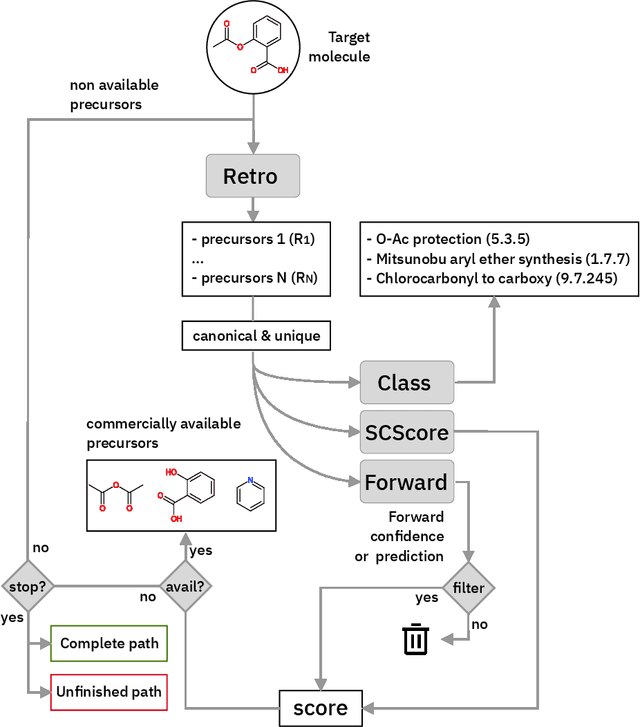
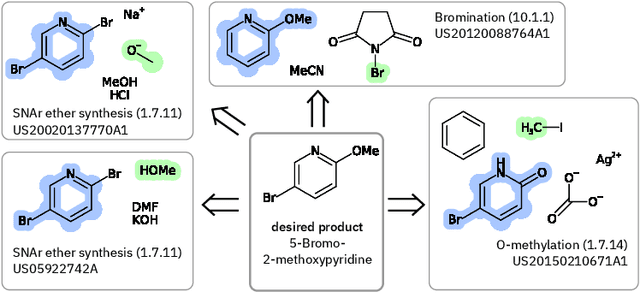
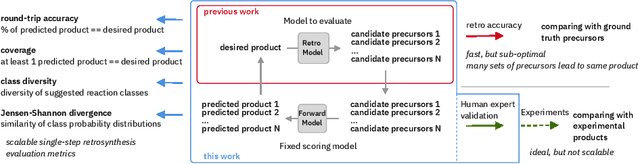
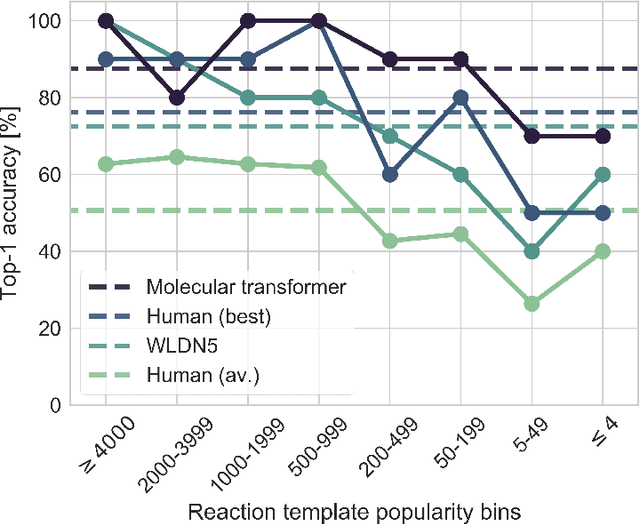
We present an extension of our Molecular Transformer architecture combined with a hyper-graph exploration strategy for automatic retrosynthesis route planning without human intervention. The single-step retrosynthetic model sets a new state of the art for predicting reactants as well as reagents, solvents and catalysts for each retrosynthetic step. We introduce new metrics (coverage, class diversity, round-trip accuracy and Jensen-Shannon divergence) to evaluate the single-step retrosynthetic models, using the forward prediction and a reaction classification model always based on the transformer architecture. The hypergraph is constructed on the fly, and the nodes are filtered and further expanded based on a Bayesian-like probability. We critically assessed the end-to-end framework with several retrosynthesis examples from literature and academic exams. Overall, the frameworks has a very good performance with few weaknesses due to the bias induced during the training process. The use of the newly introduced metrics opens up the possibility to optimize entire retrosynthetic frameworks through focusing on the performance of the single-step model only.
An Information Extraction and Knowledge Graph Platform for Accelerating Biochemical Discoveries
Jul 19, 2019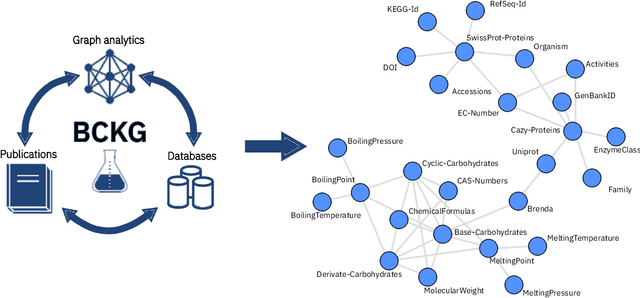
Information extraction and data mining in biochemical literature is a daunting task that demands resource-intensive computation and appropriate means to scale knowledge ingestion. Being able to leverage this immense source of technical information helps to drastically reduce costs and time to solution in multiple application fields from food safety to pharmaceutics. We present a scalable document ingestion system that integrates data from databases and publications (in PDF format) in a biochemistry knowledge graph (BCKG). The BCKG is a comprehensive source of knowledge that can be queried to retrieve known biochemical facts and to generate novel insights. After describing the knowledge ingestion framework, we showcase an application of our system in the field of carbohydrate enzymes. The BCKG represents a way to scale knowledge ingestion and automatically exploit prior knowledge to accelerate discovery in biochemical sciences.
Molecular Transformer for Chemical Reaction Prediction and Uncertainty Estimation
Nov 06, 2018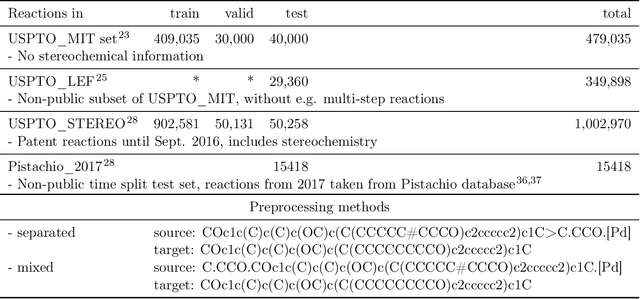
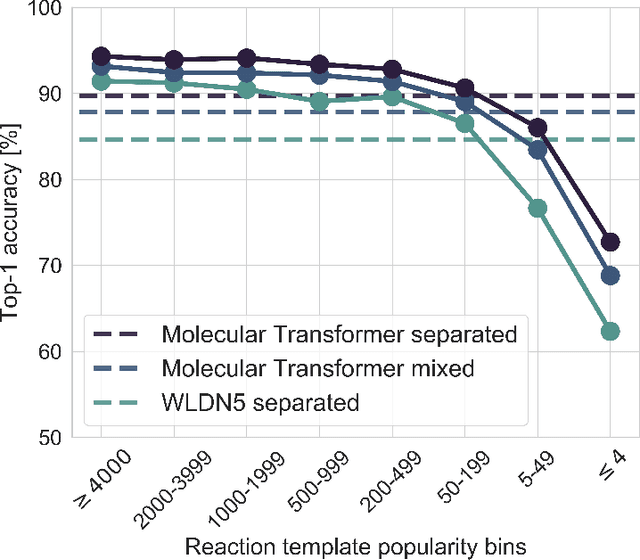
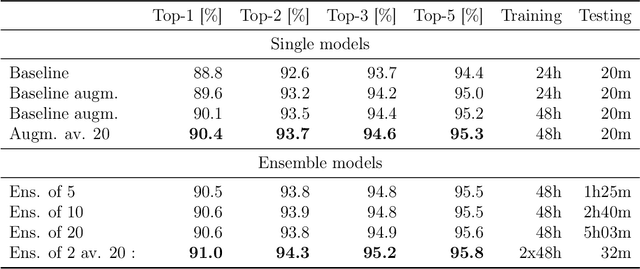
Organic synthesis is one of the key stumbling blocks in medicinal chemistry. A necessary yet unsolved step in planning synthesis is solving the forward problem: given reactants and reagents, predict the products. Similar to other works, we treat reaction prediction as a machine translation problem between SMILES strings of reactants-reagents and the products. We show that a multi-head attention Molecular Transformer model outperforms all algorithms in the literature, achieving a top-1 accuracy above 90% on a common benchmark dataset. Our algorithm requires no handcrafted rules, and accurately predicts subtle chemical transformations. Crucially, our model can accurately estimate its own uncertainty, with an uncertainty score that is 89% accurate in terms of classifying whether a prediction is correct. Furthermore, we show that the model is able to handle inputs without reactant-reagent split and including stereochemistry, which makes our method universally applicable.
"Found in Translation": Predicting Outcomes of Complex Organic Chemistry Reactions using Neural Sequence-to-Sequence Models
Nov 15, 2017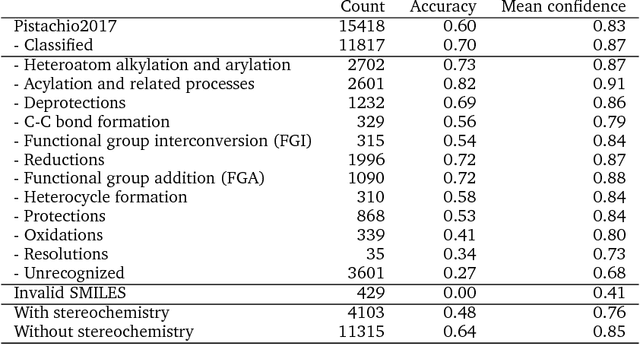
There is an intuitive analogy of an organic chemist's understanding of a compound and a language speaker's understanding of a word. Consequently, it is possible to introduce the basic concepts and analyze potential impacts of linguistic analysis to the world of organic chemistry. In this work, we cast the reaction prediction task as a translation problem by introducing a template-free sequence-to-sequence model, trained end-to-end and fully data-driven. We propose a novel way of tokenization, which is arbitrarily extensible with reaction information. With this approach, we demonstrate results superior to the state-of-the-art solution by a significant margin on the top-1 accuracy. Specifically, our approach achieves an accuracy of 80.1% without relying on auxiliary knowledge such as reaction templates. Also, 66.4% accuracy is reached on a larger and noisier dataset.