 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeExploiting Pretrained Biochemical Language Models for Targeted Drug Design
Sep 02, 2022
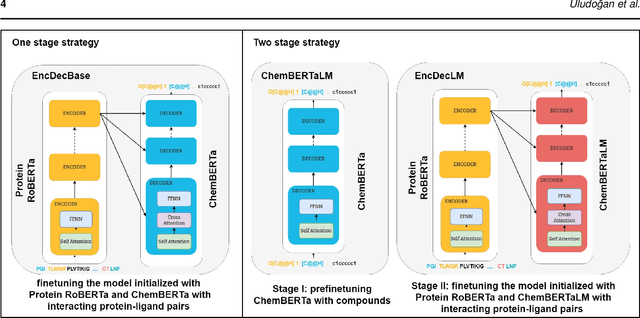

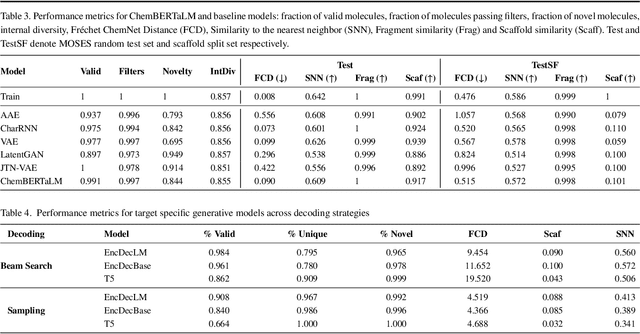
Motivation: The development of novel compounds targeting proteins of interest is one of the most important tasks in the pharmaceutical industry. Deep generative models have been applied to targeted molecular design and have shown promising results. Recently, target-specific molecule generation has been viewed as a translation between the protein language and the chemical language. However, such a model is limited by the availability of interacting protein-ligand pairs. On the other hand, large amounts of unlabeled protein sequences and chemical compounds are available and have been used to train language models that learn useful representations. In this study, we propose exploiting pretrained biochemical language models to initialize (i.e. warm start) targeted molecule generation models. We investigate two warm start strategies: (i) a one-stage strategy where the initialized model is trained on targeted molecule generation (ii) a two-stage strategy containing a pre-finetuning on molecular generation followed by target specific training. We also compare two decoding strategies to generate compounds: beam search and sampling. Results: The results show that the warm-started models perform better than a baseline model trained from scratch. The two proposed warm-start strategies achieve similar results to each other with respect to widely used metrics from benchmarks. However, docking evaluation of the generated compounds for a number of novel proteins suggests that the one-stage strategy generalizes better than the two-stage strategy. Additionally, we observe that beam search outperforms sampling in both docking evaluation and benchmark metrics for assessing compound quality. Availability and implementation: The source code is available at https://github.com/boun-tabi/biochemical-lms-for-drug-design and the materials are archived in Zenodo at https://doi.org/10.5281/zenodo.6832145
Extraction of Medication Names from Twitter Using Augmentation and an Ensemble of Language Models
Nov 12, 2021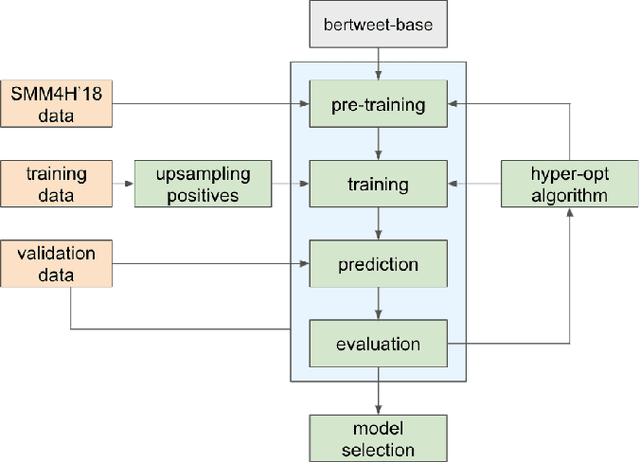
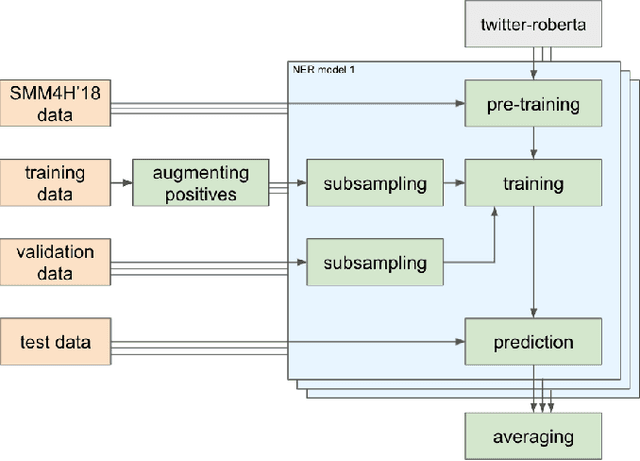
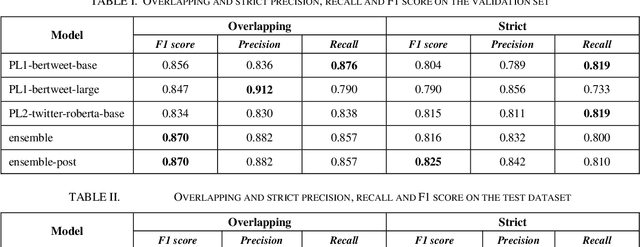
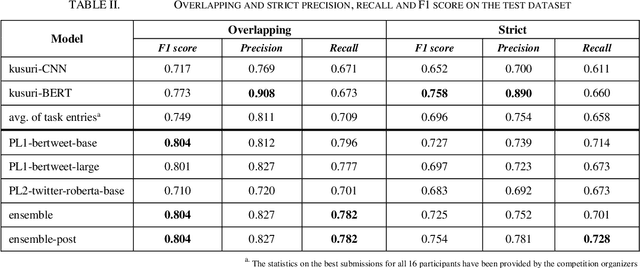
The BioCreative VII Track 3 challenge focused on the identification of medication names in Twitter user timelines. For our submission to this challenge, we expanded the available training data by using several data augmentation techniques. The augmented data was then used to fine-tune an ensemble of language models that had been pre-trained on general-domain Twitter content. The proposed approach outperformed the prior state-of-the-art algorithm Kusuri and ranked high in the competition for our selected objective function, overlapping F1 score.
Balancing Methods for Multi-label Text Classification with Long-Tailed Class Distribution
Sep 10, 2021

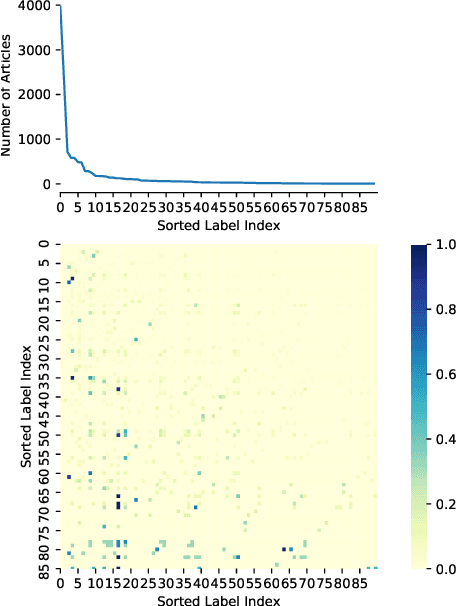
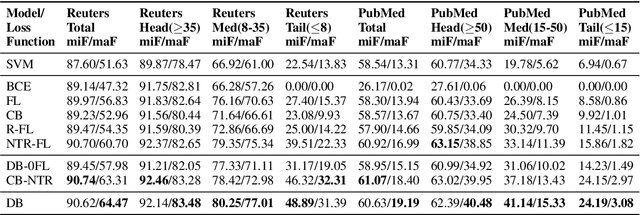
Multi-label text classification is a challenging task because it requires capturing label dependencies. It becomes even more challenging when class distribution is long-tailed. Resampling and re-weighting are common approaches used for addressing the class imbalance problem, however, they are not effective when there is label dependency besides class imbalance because they result in oversampling of common labels. Here, we introduce the application of balancing loss functions for multi-label text classification. We perform experiments on a general domain dataset with 90 labels (Reuters-21578) and a domain-specific dataset from PubMed with 18211 labels. We find that a distribution-balanced loss function, which inherently addresses both the class imbalance and label linkage problems, outperforms commonly used loss functions. Distribution balancing methods have been successfully used in the image recognition field. Here, we show their effectiveness in natural language processing. Source code is available at https://github.com/blessu/BalancedLossNLP.
Vapur: A Search Engine to Find Related Protein -- Compound Pairs in COVID-19 Literature
Sep 05, 2020Coronavirus Disease of 2019 (COVID-19) created dire consequences globally and triggered an enormous scientific effort from different domains. Resulting publications formed a gigantic domain-specific collection of text in which finding studies on a biomolecule of interest is quite challenging for general purpose search engines due to terminology-rich characteristics of the publications. Here, we present Vapur, an online COVID-19 search engine specifically designed for finding related protein - chemical pairs. Vapur is empowered with a biochemically related entities-oriented inverted index in order to group studies relevant to a biomolecule with respect to its related entities. The inverted index of Vapur is automatically created with a BioNLP pipeline and integrated with an online user interface. The online interface is designed for the smooth traversal of the current literature and is publicly available at https://tabilab.cmpe.boun.edu.tr/vapur/.
Exploring Chemical Space using Natural Language Processing Methodologies for Drug Discovery
Feb 10, 2020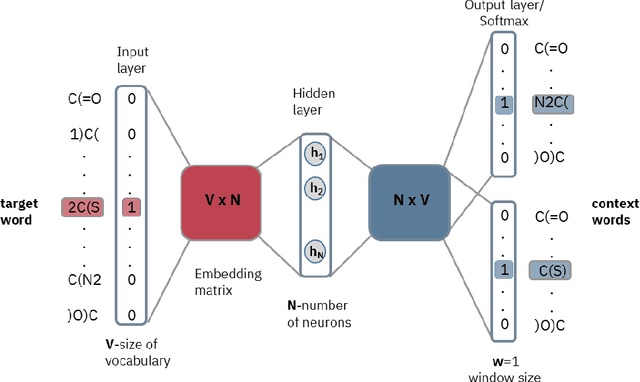
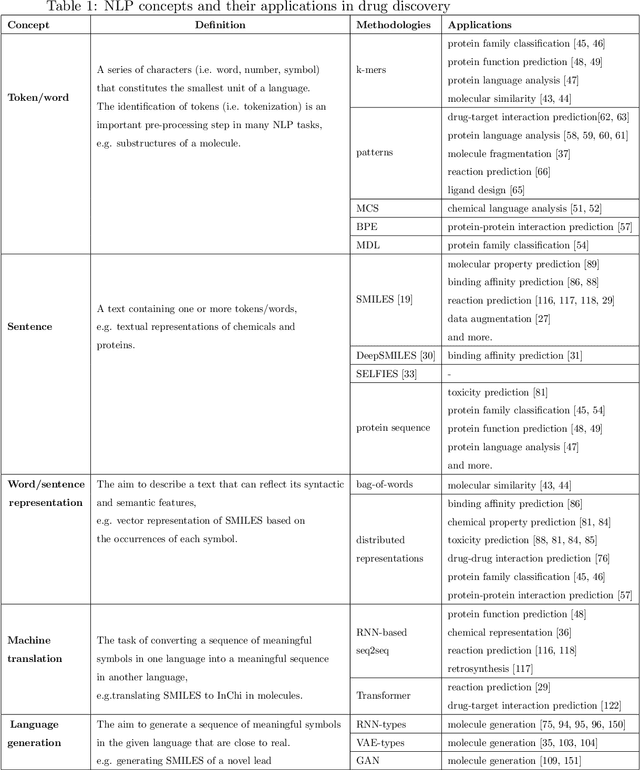
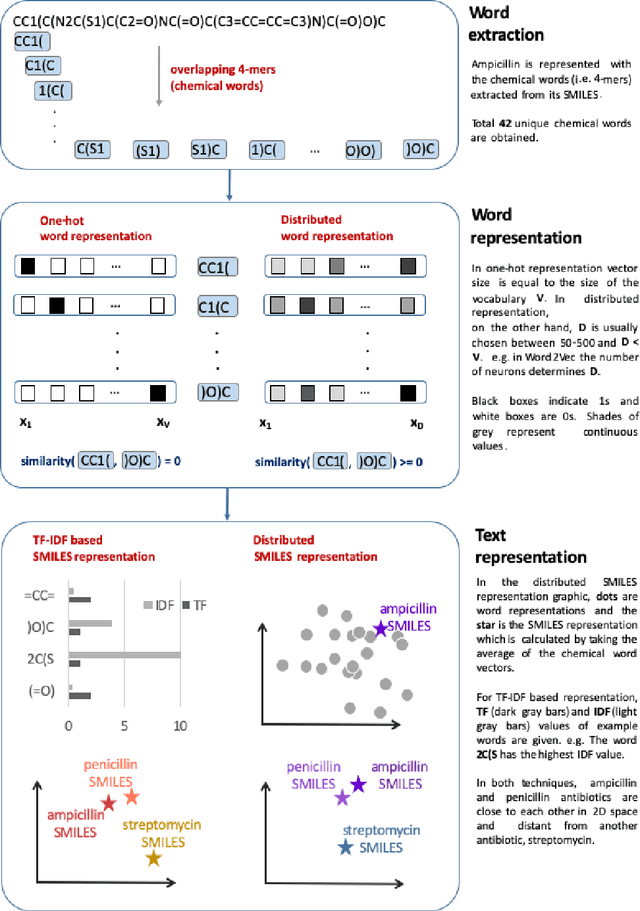
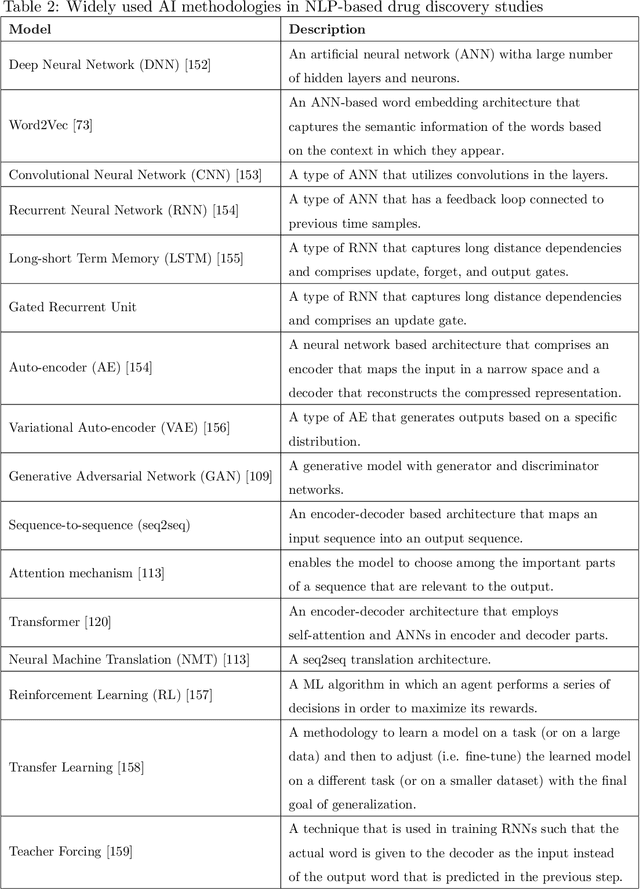
Text-based representations of chemicals and proteins can be thought of as unstructured languages codified by humans to describe domain-specific knowledge. Advances in natural language processing (NLP) methodologies in the processing of spoken languages accelerated the application of NLP to elucidate hidden knowledge in textual representations of these biochemical entities and then use it to construct models to predict molecular properties or to design novel molecules. This review outlines the impact made by these advances on drug discovery and aims to further the dialogue between medicinal chemists and computer scientists.
WideDTA: prediction of drug-target binding affinity
Feb 04, 2019
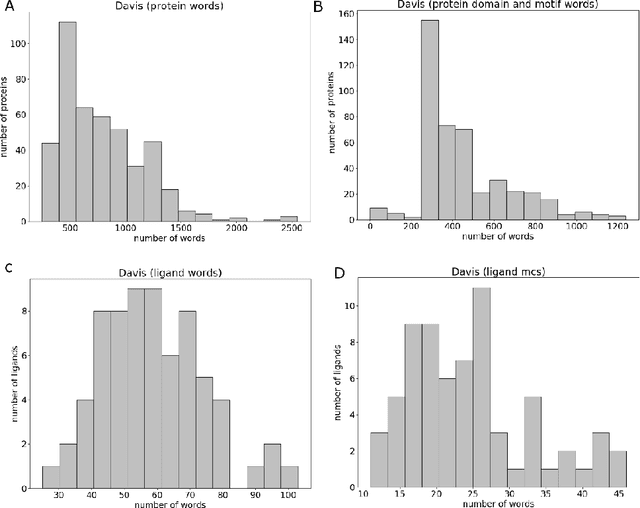
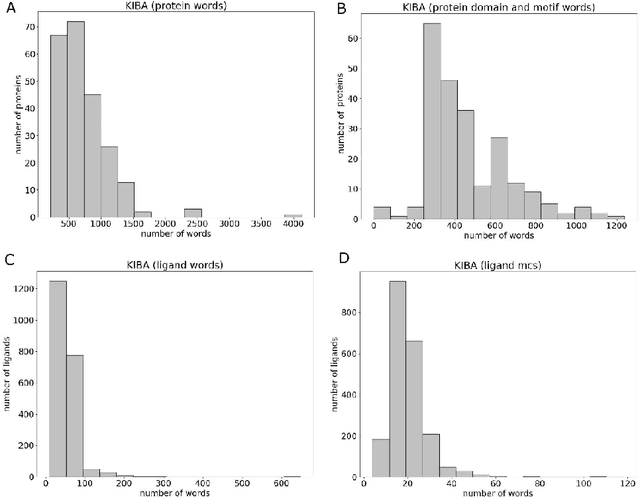
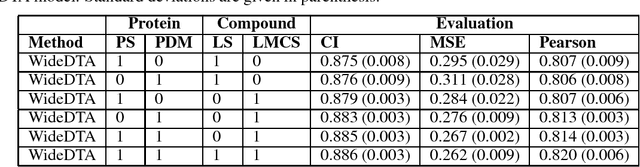
Motivation: Prediction of the interaction affinity between proteins and compounds is a major challenge in the drug discovery process. WideDTA is a deep-learning based prediction model that employs chemical and biological textual sequence information to predict binding affinity. Results: WideDTA uses four text-based information sources, namely the protein sequence, ligand SMILES, protein domains and motifs, and maximum common substructure words to predict binding affinity. WideDTA outperformed one of the state of the art deep learning methods for drug-target binding affinity prediction, DeepDTA on the KIBA dataset with a statistical significance. This indicates that the word-based sequence representation adapted by WideDTA is a promising alternative to the character-based sequence representation approach in deep learning models for binding affinity prediction, such as the one used in DeepDTA. In addition, the results showed that, given the protein sequence and ligand SMILES, the inclusion of protein domain and motif information as well as ligand maximum common substructure words do not provide additional useful information for the deep learning model. Interestingly, however, using only domain and motif information to represent proteins achieved similar performance to using the full protein sequence, suggesting that important binding relevant information is contained within the protein motifs and domains.
A chemical language based approach for protein - ligand interaction prediction
Nov 02, 2018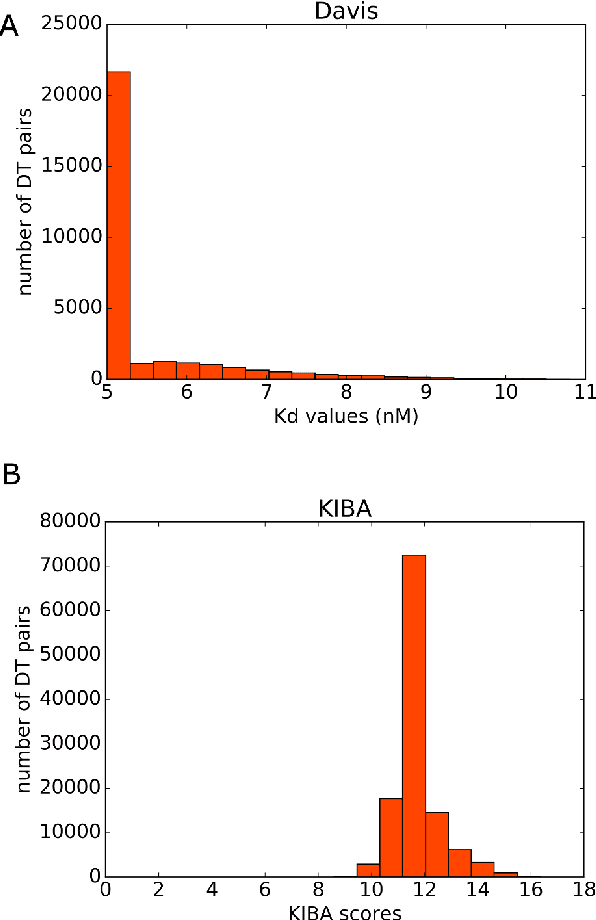
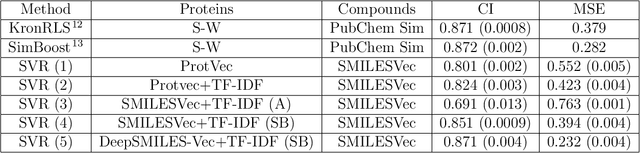
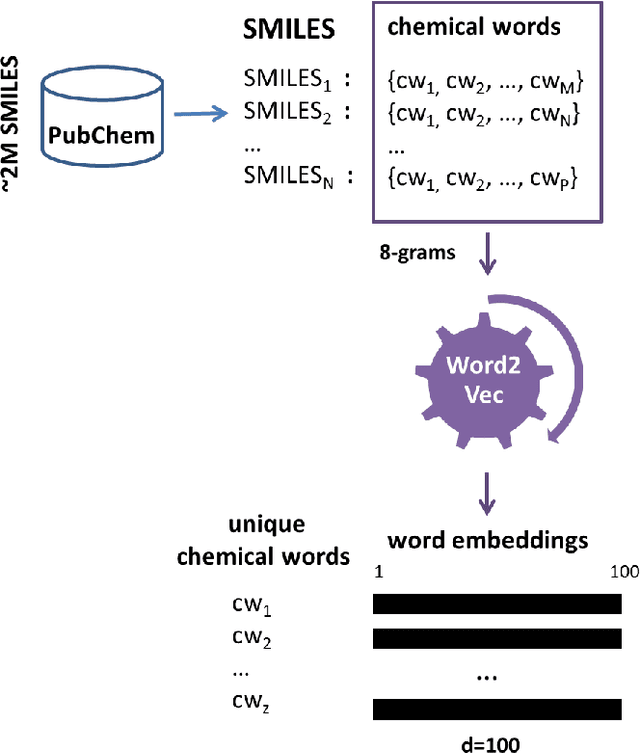
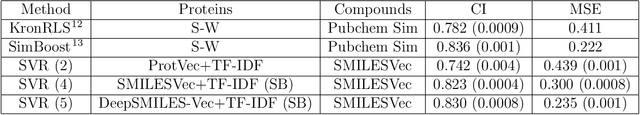
Identification of high affinity drug-target interactions (DTI) is a major research question in drug discovery. In this study, we propose a novel methodology to predict drug-target binding affinity using only ligand SMILES information. We represent proteins using the word-embeddings of the SMILES representations of their strong binding ligands. Each SMILES is represented in the form of a set of chemical words and a protein is described by the set of chemical words with the highest Term Frequency- Inverse Document Frequency (TF-IDF) value. We then utilize the Support Vector Regression (SVR) algorithm to predict protein - drug binding affinities in the Davis and KIBA Kinase datasets. We also compared the performance of SMILES representation with the recently proposed DeepSMILES representation and found that using DeepSMILES yields better performance in the prediction task. Using only SMILESVec, which is a strictly string based representation of the proteins based on their interacting ligands, we were able to predict drug-target binding affinity as well as or better than the KronRLS or SimBoost models that utilize protein sequence.
DeepDTA: Deep Drug-Target Binding Affinity Prediction
Jun 05, 2018
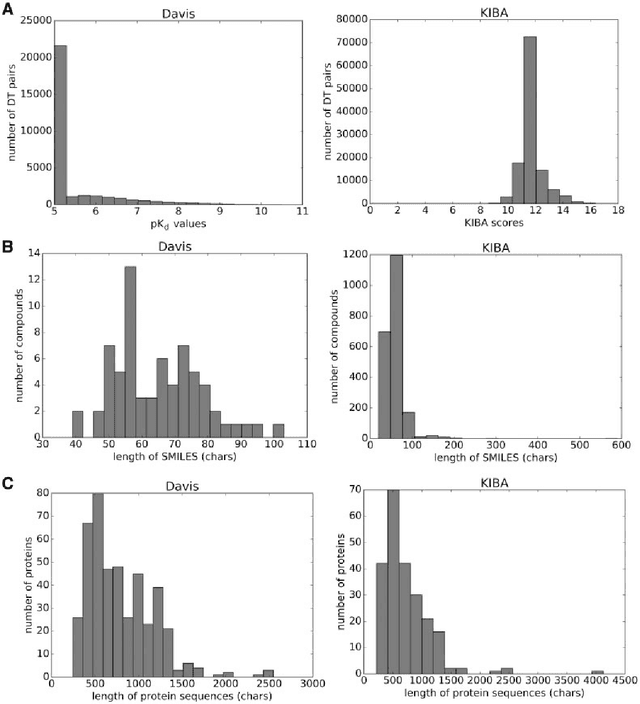
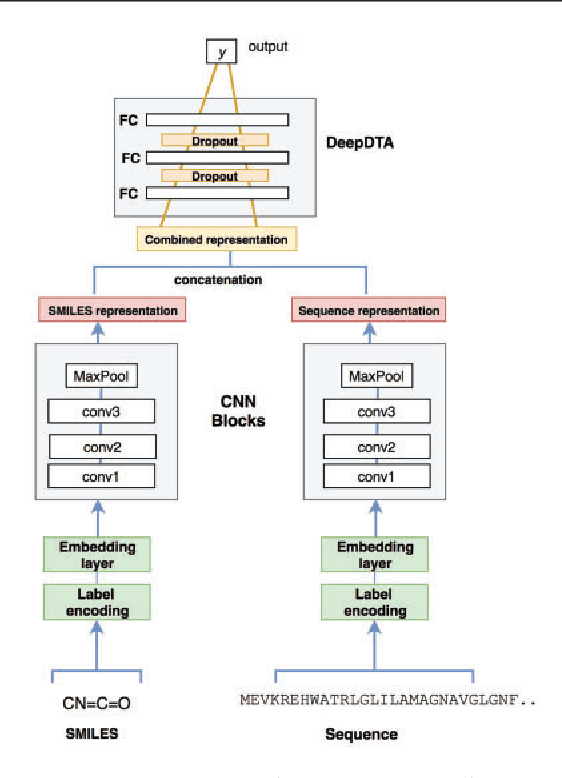

The identification of novel drug-target (DT) interactions is a substantial part of the drug discovery process. Most of the computational methods that have been proposed to predict DT interactions have focused on binary classification, where the goal is to determine whether a DT pair interacts or not. However, protein-ligand interactions assume a continuum of binding strength values, also called binding affinity and predicting this value still remains a challenge. The increase in the affinity data available in DT knowledge-bases allows the use of advanced learning techniques such as deep learning architectures in the prediction of binding affinities. In this study, we propose a deep-learning based model that uses only sequence information of both targets and drugs to predict DT interaction binding affinities. The few studies that focus on DT binding affinity prediction use either 3D structures of protein-ligand complexes or 2D features of compounds. One novel approach used in this work is the modeling of protein sequences and compound 1D representations with convolutional neural networks (CNNs). The results show that the proposed deep learning based model that uses the 1D representations of targets and drugs is an effective approach for drug target binding affinity prediction. The model in which high-level representations of a drug and a target are constructed via CNNs achieved the best Concordance Index (CI) performance in one of our larger benchmark data sets, outperforming the KronRLS algorithm and SimBoost, a state-of-the-art method for DT binding affinity prediction.
A novel methodology on distributed representations of proteins using their interacting ligands
Jan 30, 2018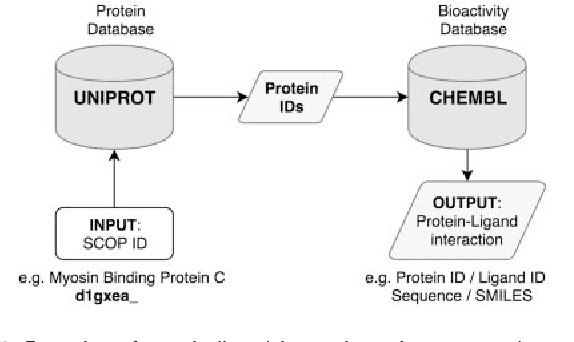

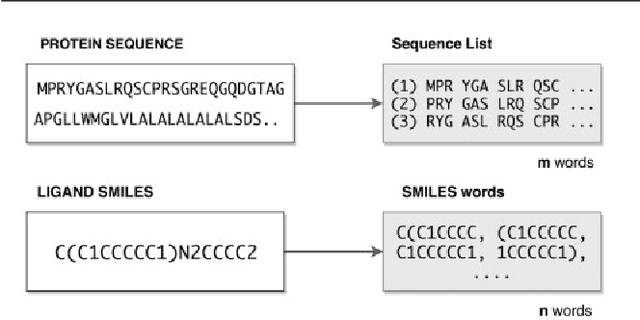
The effective representation of proteins is a crucial task that directly affects the performance of many bioinformatics problems. Related proteins usually bind to similar ligands. Chemical characteristics of ligands are known to capture the functional and mechanistic properties of proteins suggesting that a ligand based approach can be utilized in protein representation. In this study, we propose SMILESVec, a SMILES-based method to represent ligands and a novel method to compute similarity of proteins by describing them based on their ligands. The proteins are defined utilizing the word-embeddings of the SMILES strings of their ligands. The performance of the proposed protein description method is evaluated in protein clustering task using TransClust and MCL algorithms. Two other protein representation methods that utilize protein sequence, BLAST and ProtVec, and two compound fingerprint based protein representation methods are compared. We showed that ligand-based protein representation, which uses only SMILES strings of the ligands that proteins bind to, performs as well as protein-sequence based representation methods in protein clustering. The results suggest that ligand-based protein description can be an alternative to the traditional sequence or structure based representation of proteins and this novel approach can be applied to different bioinformatics problems such as prediction of new protein-ligand interactions and protein function annotation.