 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeWideDTA: prediction of drug-target binding affinity
Paper and Code

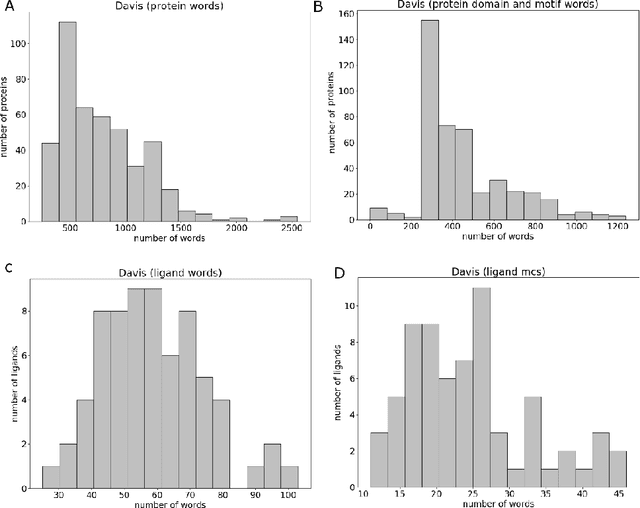
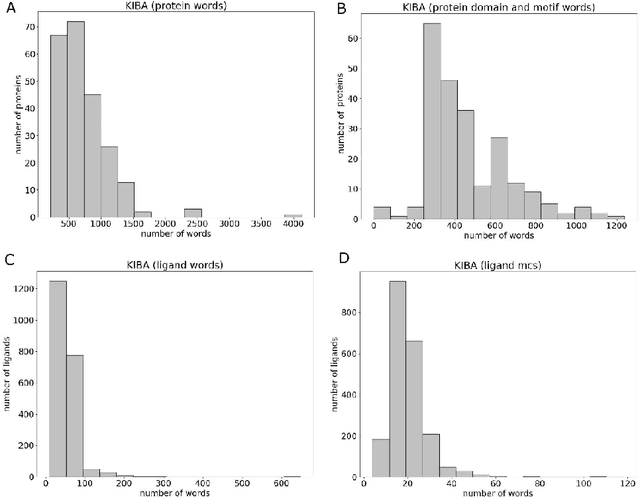
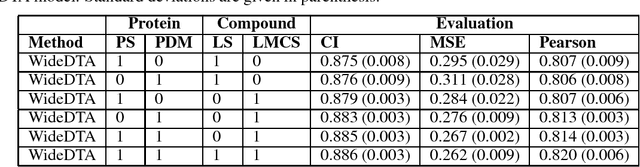
Motivation: Prediction of the interaction affinity between proteins and compounds is a major challenge in the drug discovery process. WideDTA is a deep-learning based prediction model that employs chemical and biological textual sequence information to predict binding affinity. Results: WideDTA uses four text-based information sources, namely the protein sequence, ligand SMILES, protein domains and motifs, and maximum common substructure words to predict binding affinity. WideDTA outperformed one of the state of the art deep learning methods for drug-target binding affinity prediction, DeepDTA on the KIBA dataset with a statistical significance. This indicates that the word-based sequence representation adapted by WideDTA is a promising alternative to the character-based sequence representation approach in deep learning models for binding affinity prediction, such as the one used in DeepDTA. In addition, the results showed that, given the protein sequence and ligand SMILES, the inclusion of protein domain and motif information as well as ligand maximum common substructure words do not provide additional useful information for the deep learning model. Interestingly, however, using only domain and motif information to represent proteins achieved similar performance to using the full protein sequence, suggesting that important binding relevant information is contained within the protein motifs and domains.