 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSocial and environmental impact of recent developments in machine learning on biology and chemistry research
Oct 01, 2022



Potential societal and environmental effects such as the rapidly increasing resource use and the associated environmental impact, reproducibility issues, and exclusivity, the privatization of ML research leading to a public research brain-drain, a narrowing of the research effort caused by a focus on deep learning, and the introduction of biases through a lack of sociodemographic diversity in data and personnel caused by recent developments in machine learning are a current topic of discussion and scientific publications. However, these discussions and publications focus mainly on computer science-adjacent fields, including computer vision and natural language processing or basic ML research. Using bibliometric analysis of the complete and full-text analysis of the open-access literature, we show that the same observations can be made for applied machine learning in chemistry and biology. These developments can potentially affect basic and applied research, such as drug discovery and development, beyond the known issue of biased data sets.
An automated machine learning-genetic algorithm (AutoML-GA) approach for efficient simulation-driven engine design optimization
Jan 07, 2021


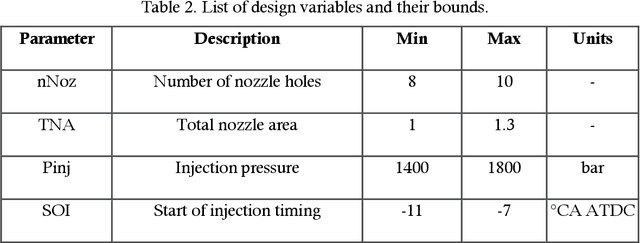
In recent years, the use of machine learning techniques as surrogate models for computational fluid dynamics (CFD) simulations has emerged as a promising method for reducing the computational cost associated with engine design optimization. However, such methods still suffer from drawbacks. One main disadvantage of such methods is that the default machine learning hyperparameters are often severely suboptimal for a given problem. This has often been addressed by manually trying out different hyperparameter settings, but this solution is ineffective in a high-dimensional hyperparameter space. Besides this problem, the amount of data needed for training is also not known a priori. In response to these issues which need to be addressed, this work describes and validates an automated active learning approach for surrogate-based optimization of internal combustion engines, AutoML-GA. In this approach, a Bayesian optimization technique is used to find the best machine learning hyperparameters based on an initial dataset obtained from a small number of CFD simulations. Subsequently, a genetic algorithm is employed to locate the design optimum on the surrogate surface trained with the optimal hyperparameters. In the vicinity of the design optimum, the solution is refined by repeatedly running CFD simulations at the projected optimum and adding the newly obtained data to the training dataset. It is shown that this approach leads to a better optimum with a lower number of CFD simulations, compared to the use of default hyperparameters. The developed approach offers the advantage of being a more hands-off approach that can be easily applied by researchers and engineers in industry who do not have a machine learning background.
Mapping the Space of Chemical Reactions Using Attention-Based Neural Networks
Dec 09, 2020
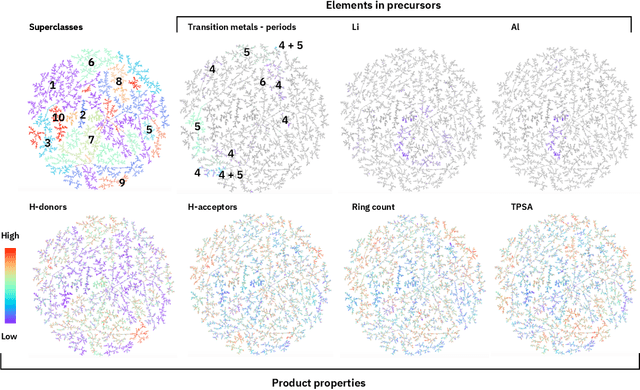
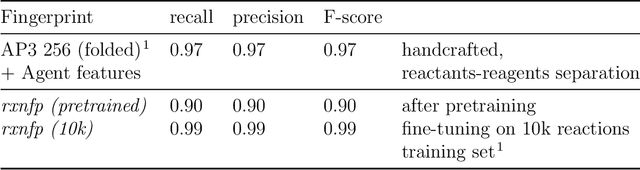

Organic reactions are usually assigned to classes containing reactions with similar reagents and mechanisms. Reaction classes facilitate the communication of complex concepts and efficient navigation through chemical reaction space. However, the classification process is a tedious task. It requires the identification of the corresponding reaction class template via annotation of the number of molecules in the reactions, the reaction center, and the distinction between reactants and reagents. This work shows that transformer-based models can infer reaction classes from non-annotated, simple text-based representations of chemical reactions. Our best model reaches a classification accuracy of 98.2%. We also show that the learned representations can be used as reaction fingerprints that capture fine-grained differences between reaction classes better than traditional reaction fingerprints. The insights into chemical reaction space enabled by our learned fingerprints are illustrated by an interactive reaction atlas providing visual clustering and similarity searching.
Visualization of Very Large High-Dimensional Data Sets as Minimum Spanning Trees
Aug 16, 2019



Here, we introduce a new data visualization and exploration method, TMAP (tree-map), which exploits locality sensitive hashing, Kruskal's minimum-spanning-tree algorithm, and a multilevel multipole-based graph layout algorithm to represent large and high dimensional data sets as a tree structure, which is readily understandable and explorable. Compared to other data visualization methods such as t-SNE or UMAP, TMAP increases the size of data sets that can be visualized due to its significantly lower memory requirements and running time and should find broad applicability in the age of big data. We exemplify TMAP in the area of cheminformatics with interactive maps for 1.16 million drug-like molecules from ChEMBL, 10.1 million small molecule fragments from FDB17, and 131 thousand 3D- structures of biomolecules from the PDB Databank, and to visualize data from literature (GUTENBERG data set), cancer biology (PANSCAN data set) and particle physics (MiniBooNE data set). TMAP is available as a Python package. Installation, usage instructions and application examples can be found at http://tmap.gdb.tools.