 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeValid and Exact Statistical Inference for Multi-dimensional Multiple Change-Points by Selective Inference
Oct 18, 2021
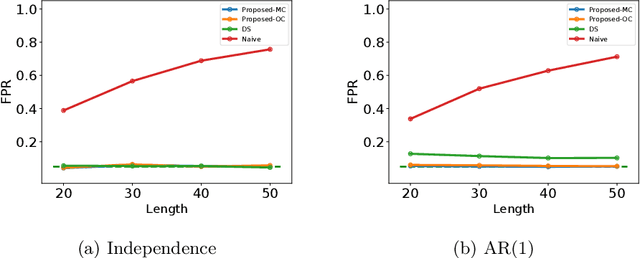
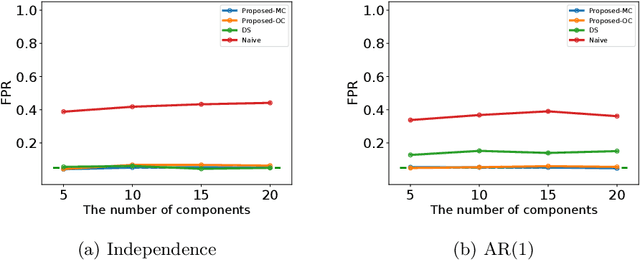

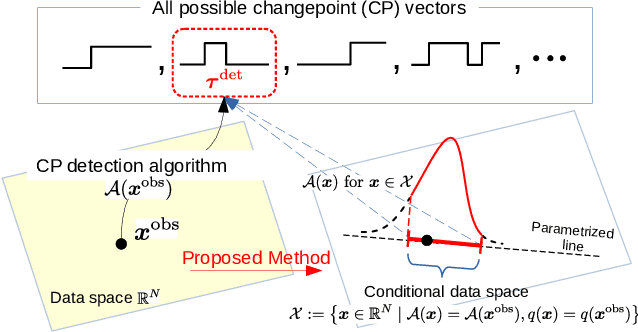
In this paper, we study statistical inference of change-points (CPs) in multi-dimensional sequence. In CP detection from a multi-dimensional sequence, it is often desirable not only to detect the location, but also to identify the subset of the components in which the change occurs. Several algorithms have been proposed for such problems, but no valid exact inference method has been established to evaluate the statistical reliability of the detected locations and components. In this study, we propose a method that can guarantee the statistical reliability of both the location and the components of the detected changes. We demonstrate the effectiveness of the proposed method by applying it to the problems of genomic abnormality identification and human behavior analysis.
Molecular Inverse-Design Platform for Material Industries
Apr 27, 2020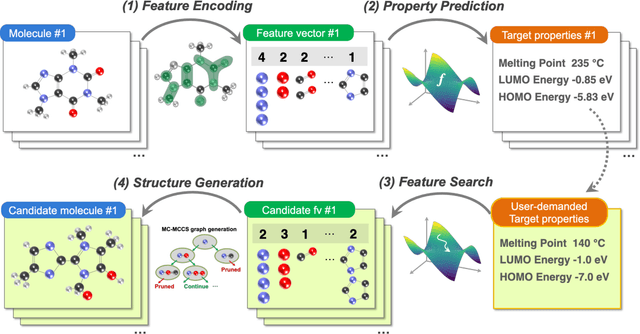
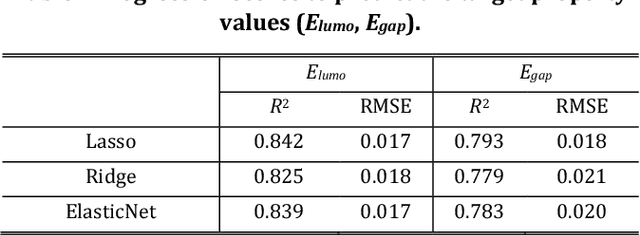
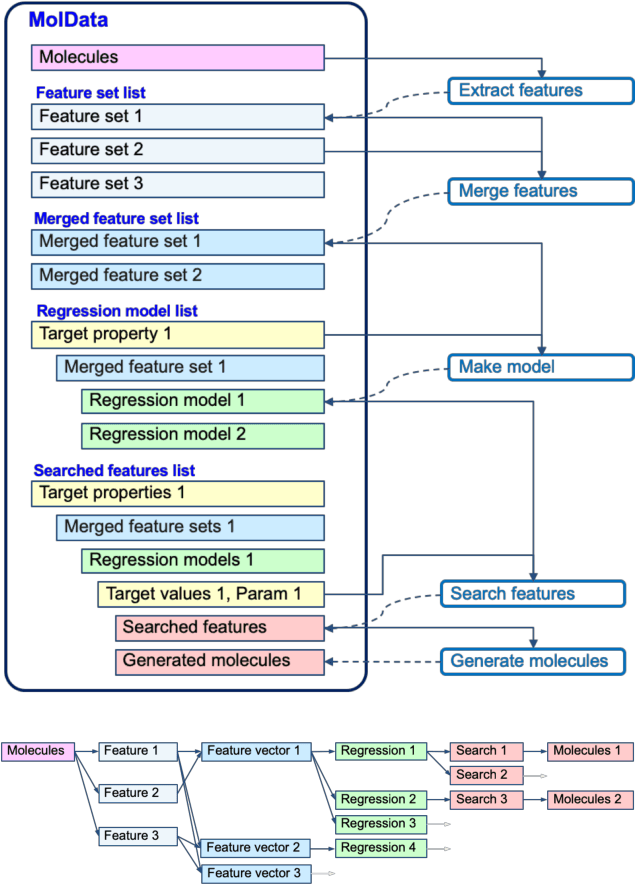
The discovery of new materials has been the essential force which brings a discontinuous improvement to industrial products' performance. However, the extra-vast combinatorial design space of material structures exceeds human experts' capability to explore all, thereby hampering material development. In this paper, we present a material industry-oriented web platform of an AI-driven molecular inverse-design system, which automatically designs brand new molecular structures rapidly and diversely. Different from existing inverse-design solutions, in this system, the combination of substructure-based feature encoding and molecular graph generation algorithms allows a user to gain high-speed, interpretable, and customizable design process. Also, a hierarchical data structure and user-oriented UI provide a flexible and intuitive workflow. The system is deployed on IBM's and our client's cloud servers and has been used by 5 partner companies. To illustrate actual industrial use cases, we exhibit inverse-design of sugar and dye molecules, that were carried out by experimental chemists in those client companies. Compared to general human chemist's standard performance, the molecular design speed was accelerated more than 10 times, and greatly increased variety was observed in the inverse-designed molecules without loss of chemical realism.
Computing Valid p-value for Optimal Changepoint by Selective Inference using Dynamic Programming
Feb 21, 2020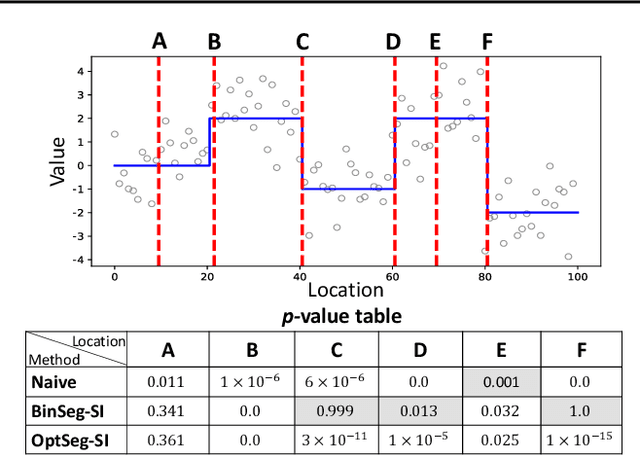
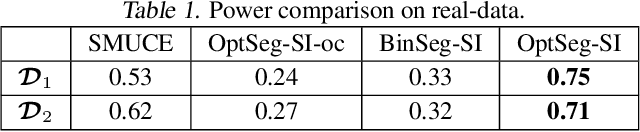
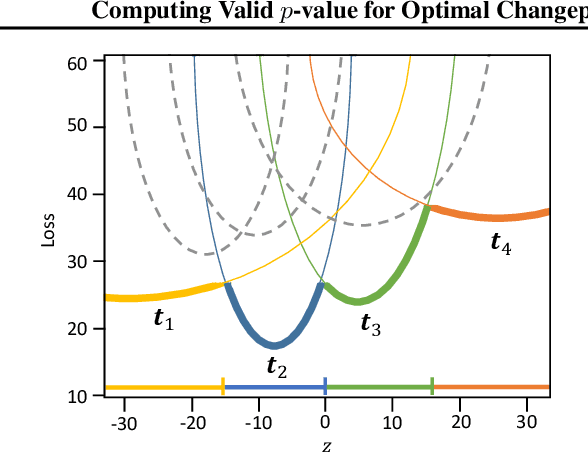
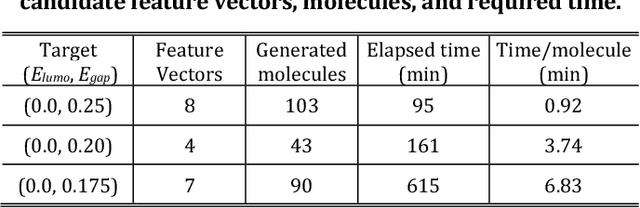
There is a vast body of literature related to methods for detecting changepoints (CP). However, less attention has been paid to assessing the statistical reliability of the detected CPs. In this paper, we introduce a novel method to perform statistical inference on the significance of the CPs, estimated by a Dynamic Programming (DP)-based optimal CP detection algorithm. Based on the selective inference (SI) framework, we propose an exact (non-asymptotic) approach to compute valid p-values for testing the significance of the CPs. Although it is well-known that SI has low statistical power because of over-conditioning, we address this disadvantage by introducing parametric programming techniques. Then, we propose an efficient method to conduct SI with the minimum amount of conditioning, leading to high statistical power. We conduct experiments on both synthetic and real-world datasets, through which we offer evidence that our proposed method is more powerful than existing methods, has decent performance in terms of computational efficiency, and provides good results in many practical applications.
An Information Extraction and Knowledge Graph Platform for Accelerating Biochemical Discoveries
Jul 19, 2019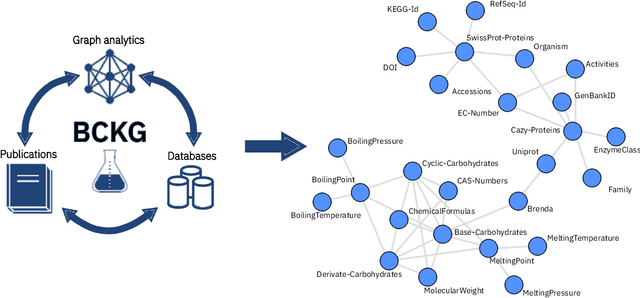
Information extraction and data mining in biochemical literature is a daunting task that demands resource-intensive computation and appropriate means to scale knowledge ingestion. Being able to leverage this immense source of technical information helps to drastically reduce costs and time to solution in multiple application fields from food safety to pharmaceutics. We present a scalable document ingestion system that integrates data from databases and publications (in PDF format) in a biochemistry knowledge graph (BCKG). The BCKG is a comprehensive source of knowledge that can be queried to retrieve known biochemical facts and to generate novel insights. After describing the knowledge ingestion framework, we showcase an application of our system in the field of carbohydrate enzymes. The BCKG represents a way to scale knowledge ingestion and automatically exploit prior knowledge to accelerate discovery in biochemical sciences.
Statistically Discriminative Sub-trajectory Mining
May 06, 2019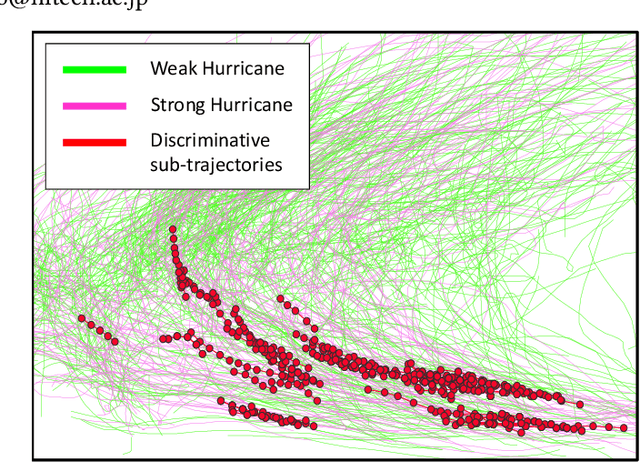
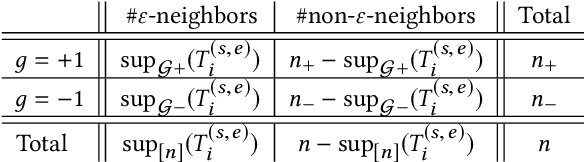
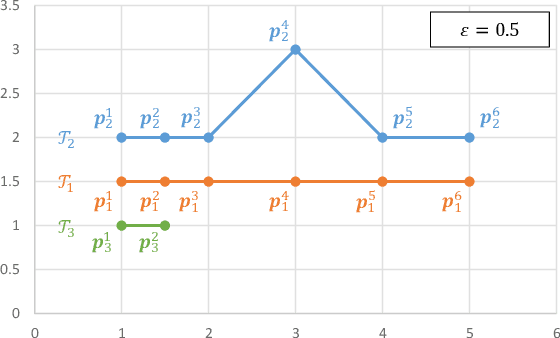
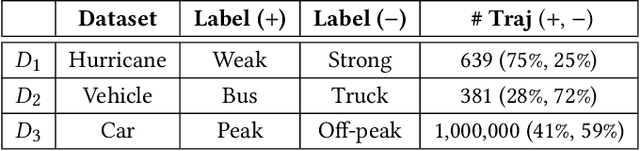
We study the problem of discriminative sub-trajectory mining. Given two groups of trajectories, the goal of this problem is to extract moving patterns in the form of sub-trajectories which are more similar to sub-trajectories of one group and less similar to those of the other. We propose a new method called Statistically Discriminative Sub-trajectory Mining (SDSM) for this problem. An advantage of the SDSM method is that the statistical significance of the extracted sub-trajectories are properly controlled in the sense that the probability of finding a false positive sub-trajectory is smaller than a specified significance threshold alpha (e.g., 0.05), which is indispensable when the method is used in scientific or social studies under noisy environment. Finding such statistically discriminative sub-trajectories from massive trajectory dataset is both computationally and statistically challenging. In the SDSM method, we resolve the difficulties by introducing a tree representation among sub-trajectories and running an efficient permutation-based statistical inference method on the tree. To the best of our knowledge, SDSM is the first method that can efficiently extract statistically discriminative sub-trajectories from massive trajectory dataset. We illustrate the effectiveness and scalability of the SDSM method by applying it to a real-world dataset with 1,000,000 trajectories which contains 16,723,602,505 sub-trajectories.