 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLLM-Fusion: A Novel Multimodal Fusion Model for Accelerated Material Discovery
Mar 02, 2025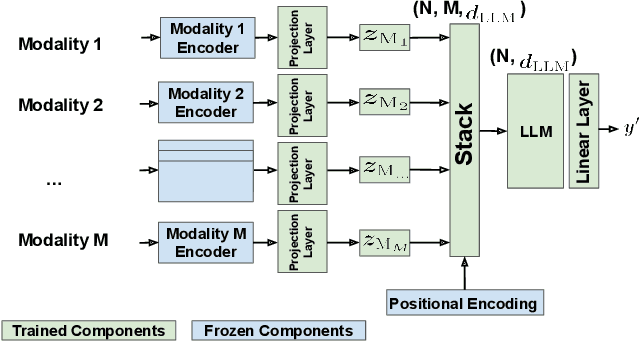
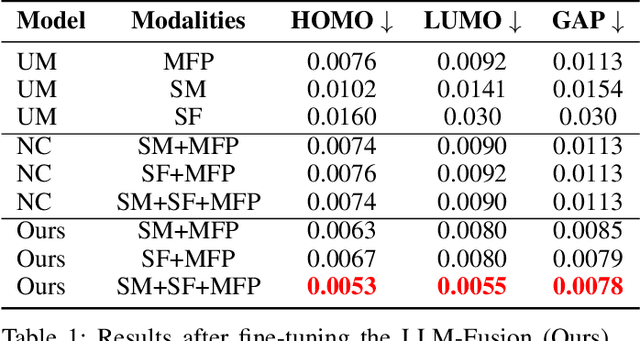
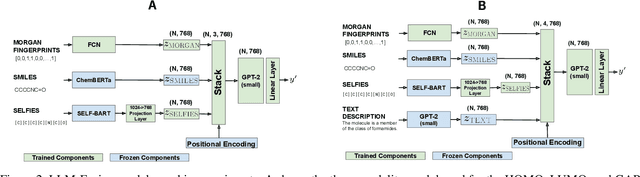

Discovering materials with desirable properties in an efficient way remains a significant problem in materials science. Many studies have tackled this problem by using different sets of information available about the materials. Among them, multimodal approaches have been found to be promising because of their ability to combine different sources of information. However, fusion algorithms to date remain simple, lacking a mechanism to provide a rich representation of multiple modalities. This paper presents LLM-Fusion, a novel multimodal fusion model that leverages large language models (LLMs) to integrate diverse representations, such as SMILES, SELFIES, text descriptions, and molecular fingerprints, for accurate property prediction. Our approach introduces a flexible LLM-based architecture that supports multimodal input processing and enables material property prediction with higher accuracy than traditional methods. We validate our model on two datasets across five prediction tasks and demonstrate its effectiveness compared to unimodal and naive concatenation baselines.
Improving Performance Prediction of Electrolyte Formulations with Transformer-based Molecular Representation Model
Jun 28, 2024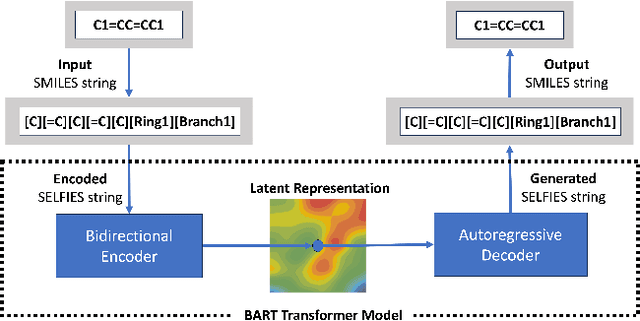

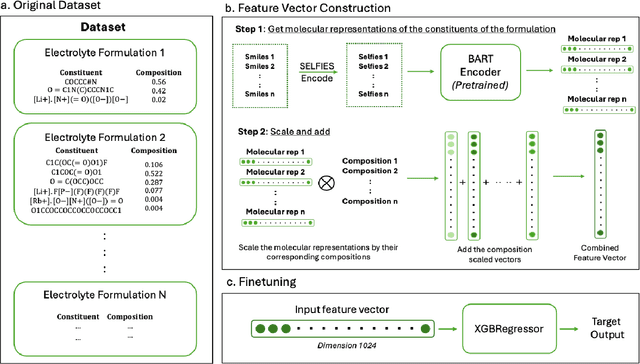

Development of efficient and high-performing electrolytes is crucial for advancing energy storage technologies, particularly in batteries. Predicting the performance of battery electrolytes rely on complex interactions between the individual constituents. Consequently, a strategy that adeptly captures these relationships and forms a robust representation of the formulation is essential for integrating with machine learning models to predict properties accurately. In this paper, we introduce a novel approach leveraging a transformer-based molecular representation model to effectively and efficiently capture the representation of electrolyte formulations. The performance of the proposed approach is evaluated on two battery property prediction tasks and the results show superior performance compared to the state-of-the-art methods.
Improving Molecular Properties Prediction Through Latent Space Fusion
Oct 20, 2023Pre-trained Language Models have emerged as promising tools for predicting molecular properties, yet their development is in its early stages, necessitating further research to enhance their efficacy and address challenges such as generalization and sample efficiency. In this paper, we present a multi-view approach that combines latent spaces derived from state-of-the-art chemical models. Our approach relies on two pivotal elements: the embeddings derived from MHG-GNN, which represent molecular structures as graphs, and MoLFormer embeddings rooted in chemical language. The attention mechanism of MoLFormer is able to identify relations between two atoms even when their distance is far apart, while the GNN of MHG-GNN can more precisely capture relations among multiple atoms closely located. In this work, we demonstrate the superior performance of our proposed multi-view approach compared to existing state-of-the-art methods, including MoLFormer-XL, which was trained on 1.1 billion molecules, particularly in intricate tasks such as predicting clinical trial drug toxicity and inhibiting HIV replication. We assessed our approach using six benchmark datasets from MoleculeNet, where it outperformed competitors in five of them. Our study highlights the potential of latent space fusion and feature integration for advancing molecular property prediction. In this work, we use small versions of MHG-GNN and MoLFormer, which opens up an opportunity for further improvement when our approach uses a larger-scale dataset.
MHG-GNN: Combination of Molecular Hypergraph Grammar with Graph Neural Network
Sep 28, 2023



Property prediction plays an important role in material discovery. As an initial step to eventually develop a foundation model for material science, we introduce a new autoencoder called the MHG-GNN, which combines graph neural network (GNN) with Molecular Hypergraph Grammar (MHG). Results on a variety of property prediction tasks with diverse materials show that MHG-GNN is promising.
GT4SD: Generative Toolkit for Scientific Discovery
Jul 08, 2022
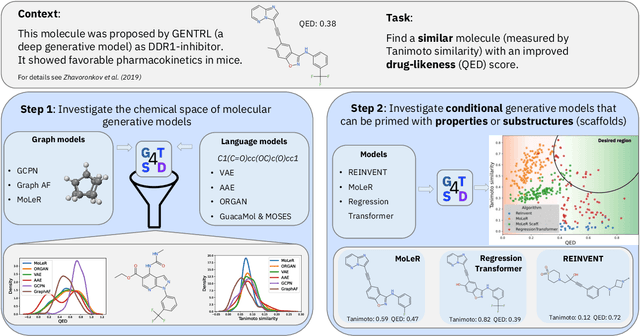
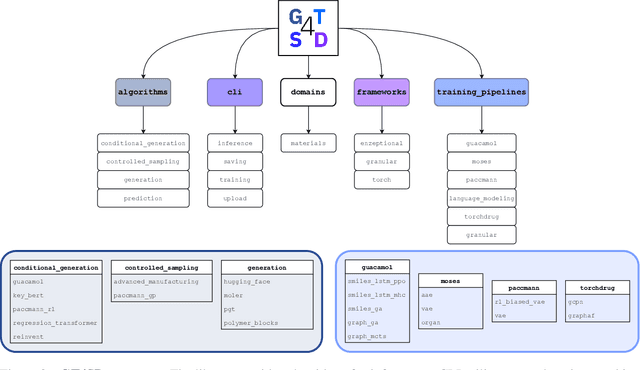
With the growing availability of data within various scientific domains, generative models hold enormous potential to accelerate scientific discovery at every step of the scientific method. Perhaps their most valuable application lies in the speeding up of what has traditionally been the slowest and most challenging step of coming up with a hypothesis. Powerful representations are now being learned from large volumes of data to generate novel hypotheses, which is making a big impact on scientific discovery applications ranging from material design to drug discovery. The GT4SD (https://github.com/GT4SD/gt4sd-core) is an extensible open-source library that enables scientists, developers and researchers to train and use state-of-the-art generative models for hypothesis generation in scientific discovery. GT4SD supports a variety of uses of generative models across material science and drug discovery, including molecule discovery and design based on properties related to target proteins, omic profiles, scaffold distances, binding energies and more.
Molecular Inverse-Design Platform for Material Industries
Apr 27, 2020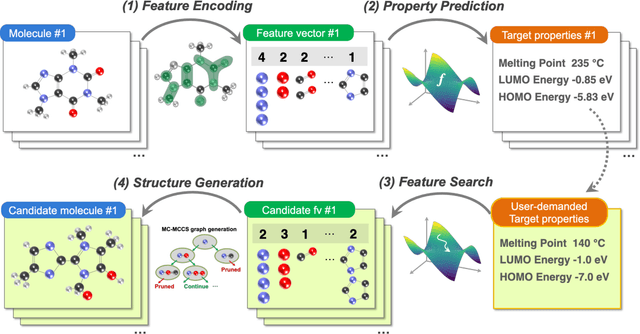
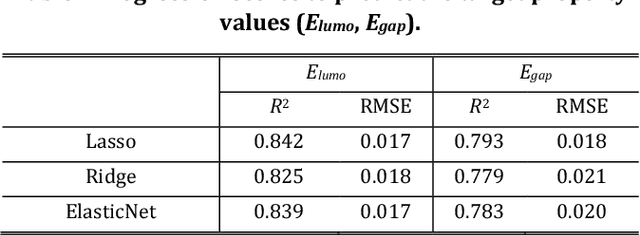
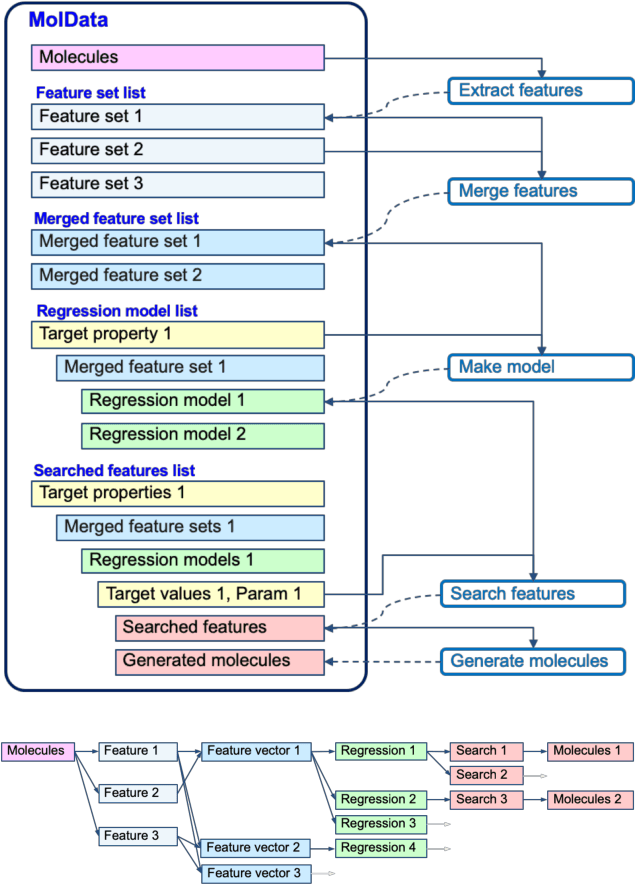
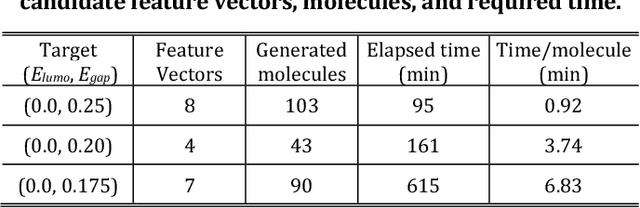
The discovery of new materials has been the essential force which brings a discontinuous improvement to industrial products' performance. However, the extra-vast combinatorial design space of material structures exceeds human experts' capability to explore all, thereby hampering material development. In this paper, we present a material industry-oriented web platform of an AI-driven molecular inverse-design system, which automatically designs brand new molecular structures rapidly and diversely. Different from existing inverse-design solutions, in this system, the combination of substructure-based feature encoding and molecular graph generation algorithms allows a user to gain high-speed, interpretable, and customizable design process. Also, a hierarchical data structure and user-oriented UI provide a flexible and intuitive workflow. The system is deployed on IBM's and our client's cloud servers and has been used by 5 partner companies. To illustrate actual industrial use cases, we exhibit inverse-design of sugar and dye molecules, that were carried out by experimental chemists in those client companies. Compared to general human chemist's standard performance, the molecular design speed was accelerated more than 10 times, and greatly increased variety was observed in the inverse-designed molecules without loss of chemical realism.