 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImproving Performance Prediction of Electrolyte Formulations with Transformer-based Molecular Representation Model
Jun 28, 2024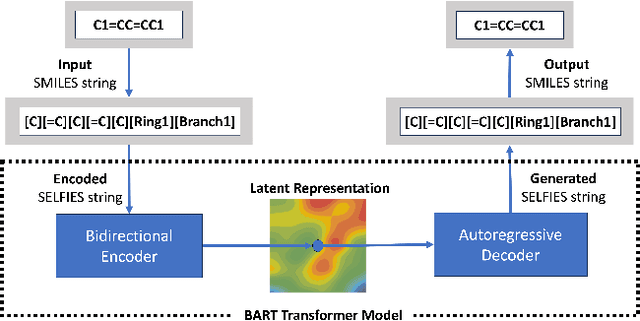

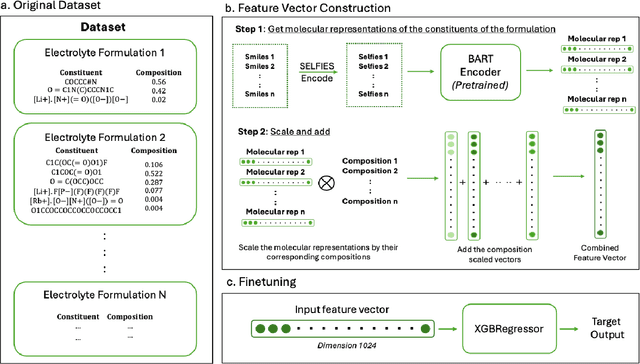

Development of efficient and high-performing electrolytes is crucial for advancing energy storage technologies, particularly in batteries. Predicting the performance of battery electrolytes rely on complex interactions between the individual constituents. Consequently, a strategy that adeptly captures these relationships and forms a robust representation of the formulation is essential for integrating with machine learning models to predict properties accurately. In this paper, we introduce a novel approach leveraging a transformer-based molecular representation model to effectively and efficiently capture the representation of electrolyte formulations. The performance of the proposed approach is evaluated on two battery property prediction tasks and the results show superior performance compared to the state-of-the-art methods.
Improving Molecular Properties Prediction Through Latent Space Fusion
Oct 20, 2023Pre-trained Language Models have emerged as promising tools for predicting molecular properties, yet their development is in its early stages, necessitating further research to enhance their efficacy and address challenges such as generalization and sample efficiency. In this paper, we present a multi-view approach that combines latent spaces derived from state-of-the-art chemical models. Our approach relies on two pivotal elements: the embeddings derived from MHG-GNN, which represent molecular structures as graphs, and MoLFormer embeddings rooted in chemical language. The attention mechanism of MoLFormer is able to identify relations between two atoms even when their distance is far apart, while the GNN of MHG-GNN can more precisely capture relations among multiple atoms closely located. In this work, we demonstrate the superior performance of our proposed multi-view approach compared to existing state-of-the-art methods, including MoLFormer-XL, which was trained on 1.1 billion molecules, particularly in intricate tasks such as predicting clinical trial drug toxicity and inhibiting HIV replication. We assessed our approach using six benchmark datasets from MoleculeNet, where it outperformed competitors in five of them. Our study highlights the potential of latent space fusion and feature integration for advancing molecular property prediction. In this work, we use small versions of MHG-GNN and MoLFormer, which opens up an opportunity for further improvement when our approach uses a larger-scale dataset.
MHG-GNN: Combination of Molecular Hypergraph Grammar with Graph Neural Network
Sep 28, 2023



Property prediction plays an important role in material discovery. As an initial step to eventually develop a foundation model for material science, we introduce a new autoencoder called the MHG-GNN, which combines graph neural network (GNN) with Molecular Hypergraph Grammar (MHG). Results on a variety of property prediction tasks with diverse materials show that MHG-GNN is promising.
An Ensemble Approach for Automated Theorem Proving Based on Efficient Name Invariant Graph Neural Representations
May 15, 2023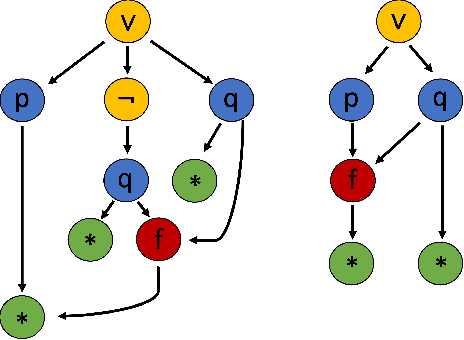
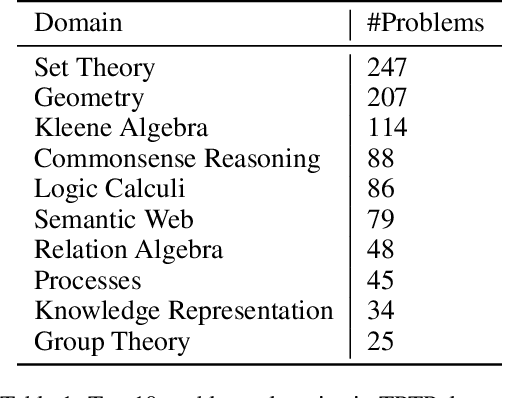
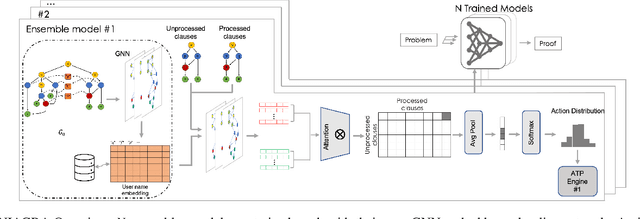
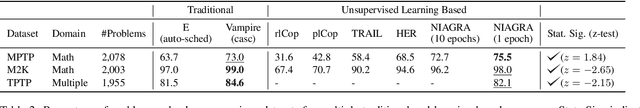
Using reinforcement learning for automated theorem proving has recently received much attention. Current approaches use representations of logical statements that often rely on the names used in these statements and, as a result, the models are generally not transferable from one domain to another. The size of these representations and whether to include the whole theory or part of it are other important decisions that affect the performance of these approaches as well as their runtime efficiency. In this paper, we present NIAGRA; an ensemble Name InvAriant Graph RepresentAtion. NIAGRA addresses this problem by using 1) improved Graph Neural Networks for learning name-invariant formula representations that is tailored for their unique characteristics and 2) an efficient ensemble approach for automated theorem proving. Our experimental evaluation shows state-of-the-art performance on multiple datasets from different domains with improvements up to 10% compared to the best learning-based approaches. Furthermore, transfer learning experiments show that our approach significantly outperforms other learning-based approaches by up to 28%.
GT4SD: Generative Toolkit for Scientific Discovery
Jul 08, 2022
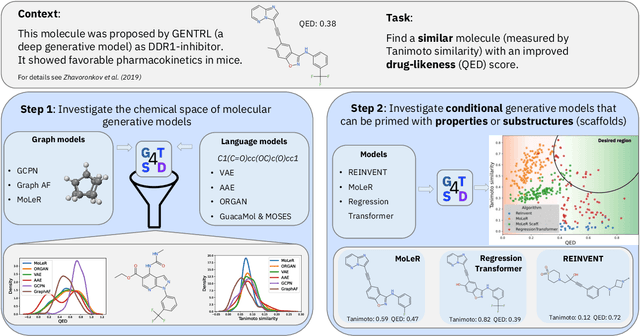
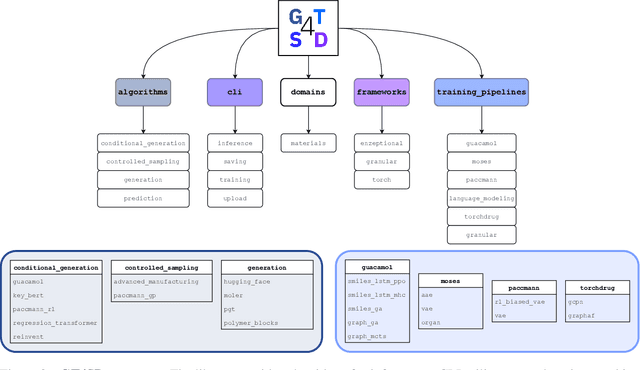
With the growing availability of data within various scientific domains, generative models hold enormous potential to accelerate scientific discovery at every step of the scientific method. Perhaps their most valuable application lies in the speeding up of what has traditionally been the slowest and most challenging step of coming up with a hypothesis. Powerful representations are now being learned from large volumes of data to generate novel hypotheses, which is making a big impact on scientific discovery applications ranging from material design to drug discovery. The GT4SD (https://github.com/GT4SD/gt4sd-core) is an extensible open-source library that enables scientists, developers and researchers to train and use state-of-the-art generative models for hypothesis generation in scientific discovery. GT4SD supports a variety of uses of generative models across material science and drug discovery, including molecule discovery and design based on properties related to target proteins, omic profiles, scaffold distances, binding energies and more.
Designing Machine Learning Pipeline Toolkit for AutoML Surrogate Modeling Optimization
Jul 14, 2021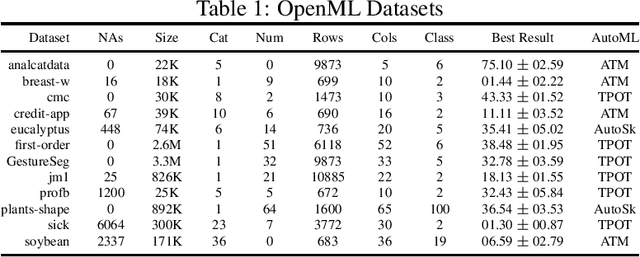
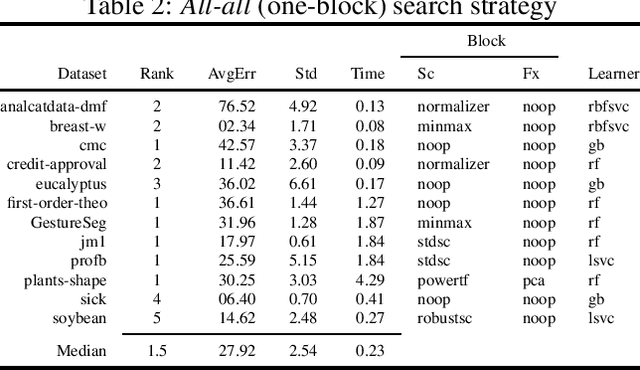
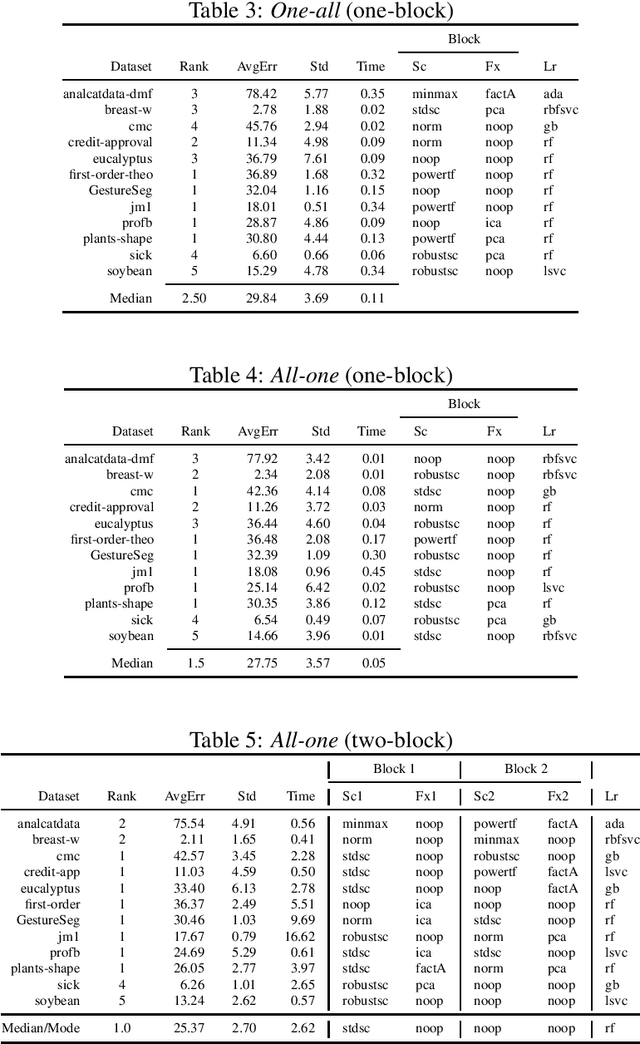
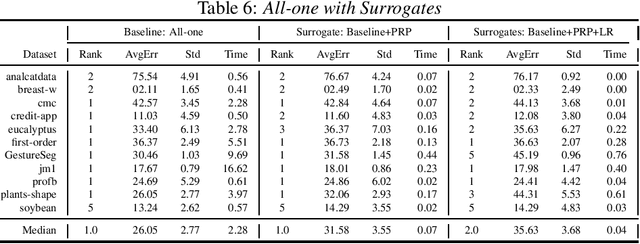
The pipeline optimization problem in machine learning requires simultaneous optimization of pipeline structures and parameter adaptation of their elements. Having an elegant way to express these structures can help lessen the complexity in the management and analysis of their performances together with the different choices of optimization strategies. With these issues in mind, we created the AutoMLPipeline (AMLP) toolkit which facilitates the creation and evaluation of complex machine learning pipeline structures using simple expressions. We use AMLP to find optimal pipeline signatures, datamine them, and use these datamined features to speed-up learning and prediction. We formulated a two-stage pipeline optimization with surrogate modeling in AMLP which outperforms other AutoML approaches with a 4-hour time budget in less than 5 minutes of AMLP computation time.
Estimating Blink Probability for Highlight Detection in Figure Skating Videos
Jul 02, 2020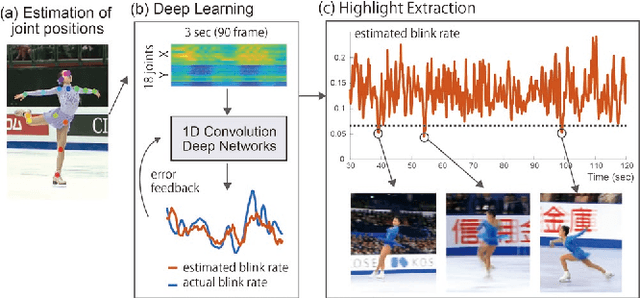
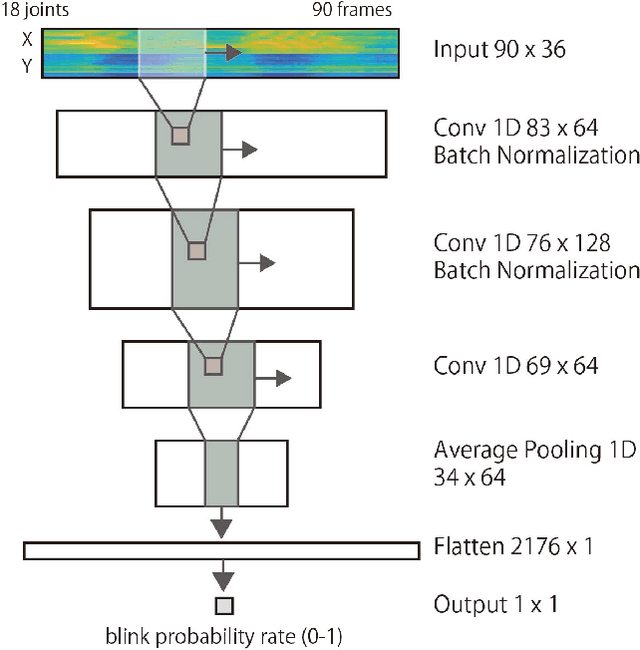
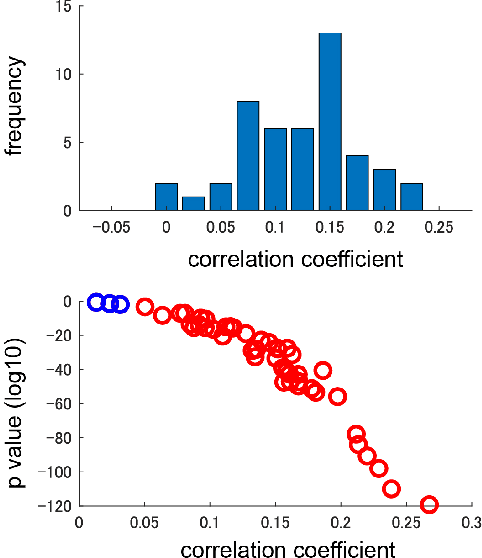
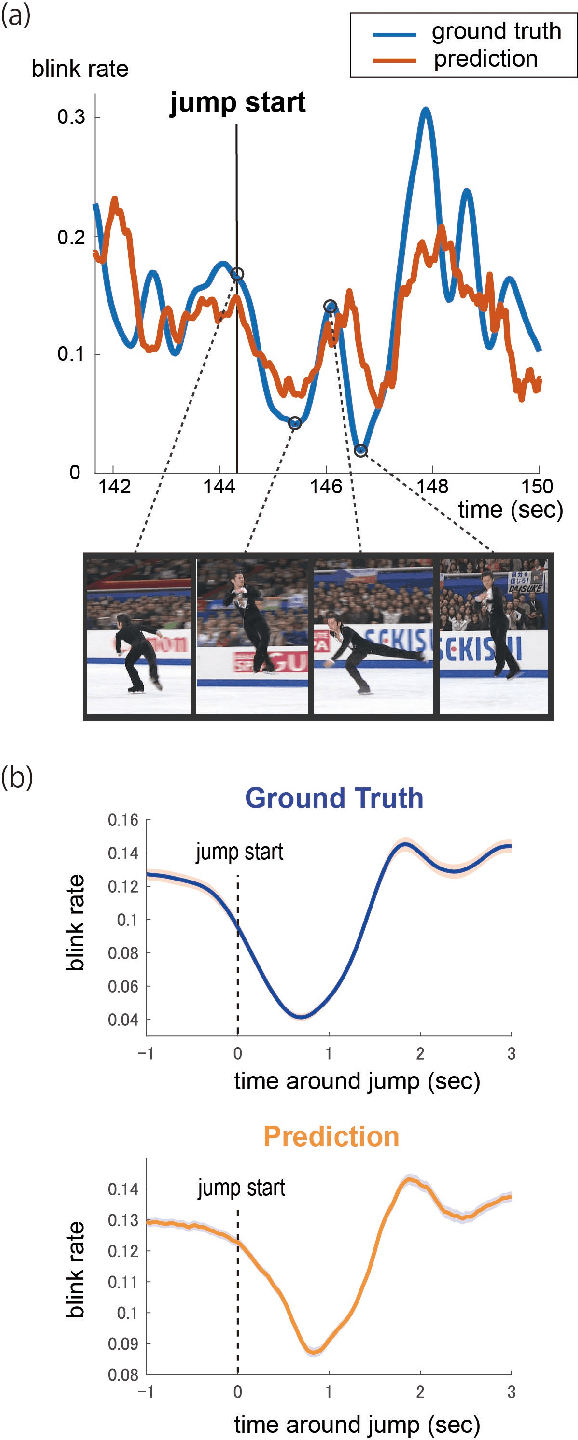
Highlight detection in sports videos has a broad viewership and huge commercial potential. It is thus imperative to detect highlight scenes more suitably for human interest with high temporal accuracy. Since people instinctively suppress blinks during attention-grabbing events and synchronously generate blinks at attention break points in videos, the instantaneous blink rate can be utilized as a highly accurate temporal indicator of human interest. Therefore, in this study, we propose a novel, automatic highlight detection method based on the blink rate. The method trains a one-dimensional convolution network (1D-CNN) to assess blink rates at each video frame from the spatio-temporal pose features of figure skating videos. Experiments show that the method successfully estimates the blink rate in 94% of the video clips and predicts the temporal change in the blink rate around a jump event with high accuracy. Moreover, the method detects not only the representative athletic action, but also the distinctive artistic expression of figure skating performance as key frames. This suggests that the blink-rate-based supervised learning approach enables high-accuracy highlight detection that more closely matches human sensibility.
Generating Dialogue Agents via Automated Planning
Feb 02, 2019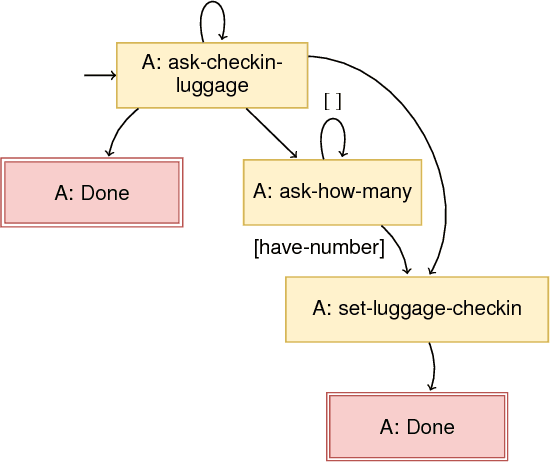
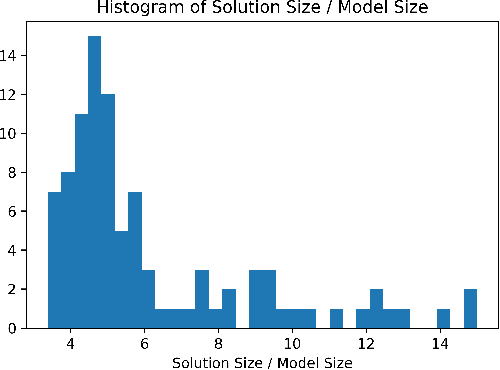
Dialogue systems have many applications such as customer support or question answering. Typically they have been limited to shallow single turn interactions. However more advanced applications such as career coaching or planning a trip require a much more complex multi-turn dialogue. Current limitations of conversational systems have made it difficult to support applications that require personalization, customization and context dependent interactions. We tackle this challenging problem by using domain-independent AI planning to automatically create dialogue plans, customized to guide a dialogue towards achieving a given goal. The input includes a library of atomic dialogue actions, an initial state of the dialogue, and a goal. Dialogue plans are plugged into a dialogue system capable to orchestrate their execution. Use cases demonstrate the viability of the approach. Our work on dialogue planning has been integrated into a product, and it is in the process of being deployed into another.
A Survey of Parallel A*
Aug 16, 2017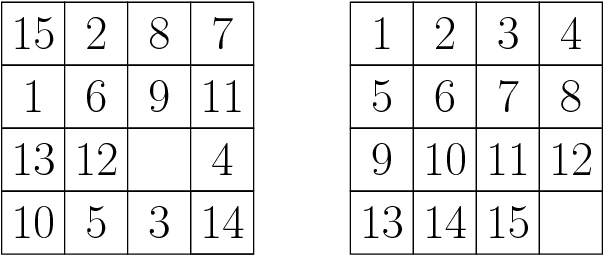
A* is a best-first search algorithm for finding optimal-cost paths in graphs. A* benefits significantly from parallelism because in many applications, A* is limited by memory usage, so distributed memory implementations of A* that use all of the aggregate memory on the cluster enable problems that can not be solved by serial, single-machine implementations to be solved. We survey approaches to parallel A*, focusing on decentralized approaches to A* which partition the state space among processors. We also survey approaches to parallel, limited-memory variants of A* such as parallel IDA*.
Evaluation of a Simple, Scalable, Parallel Best-First Search Strategy
Oct 25, 2012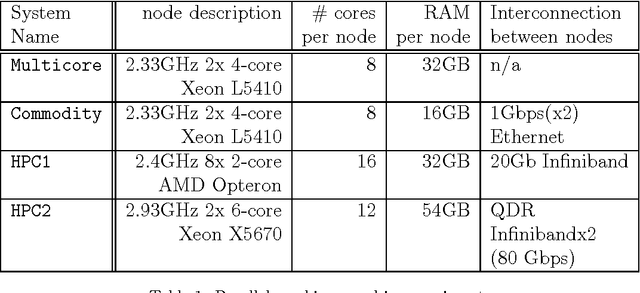
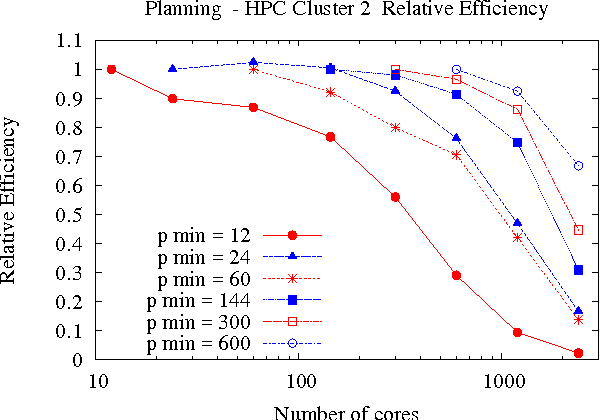
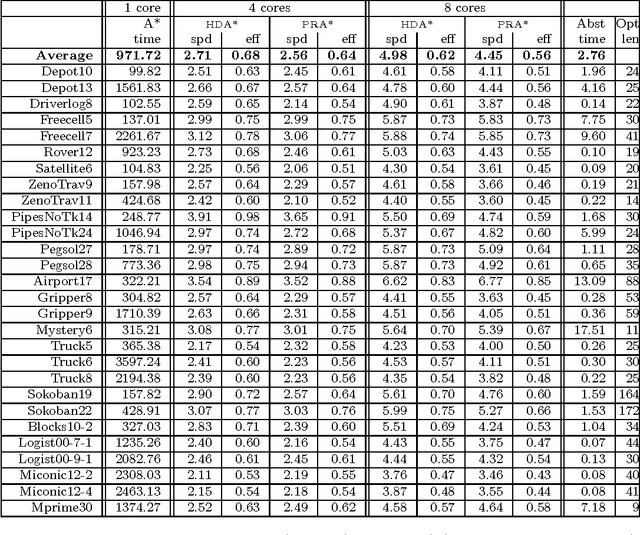
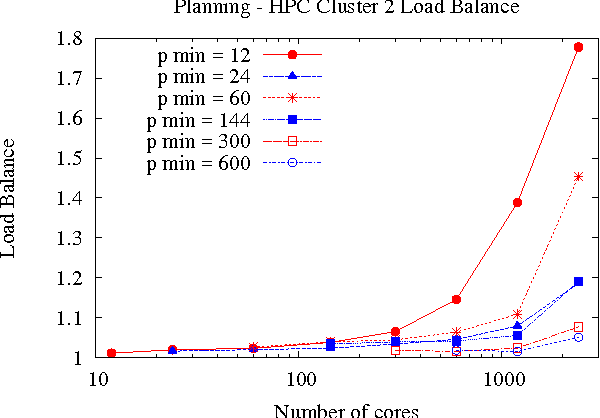
Large-scale, parallel clusters composed of commodity processors are increasingly available, enabling the use of vast processing capabilities and distributed RAM to solve hard search problems. We investigate Hash-Distributed A* (HDA*), a simple approach to parallel best-first search that asynchronously distributes and schedules work among processors based on a hash function of the search state. We use this approach to parallelize the A* algorithm in an optimal sequential version of the Fast Downward planner, as well as a 24-puzzle solver. The scaling behavior of HDA* is evaluated experimentally on a shared memory, multicore machine with 8 cores, a cluster of commodity machines using up to 64 cores, and large-scale high-performance clusters, using up to 2400 processors. We show that this approach scales well, allowing the effective utilization of large amounts of distributed memory to optimally solve problems which require terabytes of RAM. We also compare HDA* to Transposition-table Driven Scheduling (TDS), a hash-based parallelization of IDA*, and show that, in planning, HDA* significantly outperforms TDS. A simple hybrid which combines HDA* and TDS to exploit strengths of both algorithms is proposed and evaluated.
* in press, to appear in Artificial Intelligence