 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAutomated, LLM enabled extraction of synthesis details for reticular materials from scientific literature
Nov 05, 2024



Automated knowledge extraction from scientific literature can potentially accelerate materials discovery. We have investigated an approach for extracting synthesis protocols for reticular materials from scientific literature using large language models (LLMs). To that end, we introduce a Knowledge Extraction Pipeline (KEP) that automatizes LLM-assisted paragraph classification and information extraction. By applying prompt engineering with in-context learning (ICL) to a set of open-source LLMs, we demonstrate that LLMs can retrieve chemical information from PDF documents, without the need for fine-tuning or training and at a reduced risk of hallucination. By comparing the performance of five open-source families of LLMs in both paragraph classification and information extraction tasks, we observe excellent model performance even if only few example paragraphs are included in the ICL prompts. The results show the potential of the KEP approach for reducing human annotations and data curation efforts in automated scientific knowledge extraction.
A Large Encoder-Decoder Family of Foundation Models For Chemical Language
Jul 24, 2024Large-scale pre-training methodologies for chemical language models represent a breakthrough in cheminformatics. These methods excel in tasks such as property prediction and molecule generation by learning contextualized representations of input tokens through self-supervised learning on large unlabeled corpora. Typically, this involves pre-training on unlabeled data followed by fine-tuning on specific tasks, reducing dependence on annotated datasets and broadening chemical language representation understanding. This paper introduces a large encoder-decoder chemical foundation models pre-trained on a curated dataset of 91 million SMILES samples sourced from PubChem, which is equivalent to 4 billion of molecular tokens. The proposed foundation model supports different complex tasks, including quantum property prediction, and offer flexibility with two main variants (289M and $8\times289M$). Our experiments across multiple benchmark datasets validate the capacity of the proposed model in providing state-of-the-art results for different tasks. We also provide a preliminary assessment of the compositionality of the embedding space as a prerequisite for the reasoning tasks. We demonstrate that the produced latent space is separable compared to the state-of-the-art with few-shot learning capabilities.
KIF: A Framework for Virtual Integration of Heterogeneous Knowledge Bases using Wikidata
Mar 15, 2024We present a knowledge integration framework (called KIF) that uses Wikidata as a lingua franca to integrate heterogeneous knowledge bases. These can be triplestores, relational databases, CSV files, etc., which may or may not use the Wikidata dialect of RDF. KIF leverages Wikidata's data model and vocabulary plus user-defined mappings to expose a unified view of the integrated bases while keeping track of the context and provenance of their statements. The result is a virtual knowledge base which behaves like an "extended Wikidata" and which can be queried either through an efficient filter interface or using SPARQL. We present the design and implementation of KIF, discuss how we have used it to solve a real integration problem in the domain of chemistry (involving Wikidata, PubChem, and IBM CIRCA), and present experimental results on the performance and overhead of KIF.
Improving Molecular Properties Prediction Through Latent Space Fusion
Oct 20, 2023Pre-trained Language Models have emerged as promising tools for predicting molecular properties, yet their development is in its early stages, necessitating further research to enhance their efficacy and address challenges such as generalization and sample efficiency. In this paper, we present a multi-view approach that combines latent spaces derived from state-of-the-art chemical models. Our approach relies on two pivotal elements: the embeddings derived from MHG-GNN, which represent molecular structures as graphs, and MoLFormer embeddings rooted in chemical language. The attention mechanism of MoLFormer is able to identify relations between two atoms even when their distance is far apart, while the GNN of MHG-GNN can more precisely capture relations among multiple atoms closely located. In this work, we demonstrate the superior performance of our proposed multi-view approach compared to existing state-of-the-art methods, including MoLFormer-XL, which was trained on 1.1 billion molecules, particularly in intricate tasks such as predicting clinical trial drug toxicity and inhibiting HIV replication. We assessed our approach using six benchmark datasets from MoleculeNet, where it outperformed competitors in five of them. Our study highlights the potential of latent space fusion and feature integration for advancing molecular property prediction. In this work, we use small versions of MHG-GNN and MoLFormer, which opens up an opportunity for further improvement when our approach uses a larger-scale dataset.
Beyond Chemical Language: A Multimodal Approach to Enhance Molecular Property Prediction
Jun 22, 2023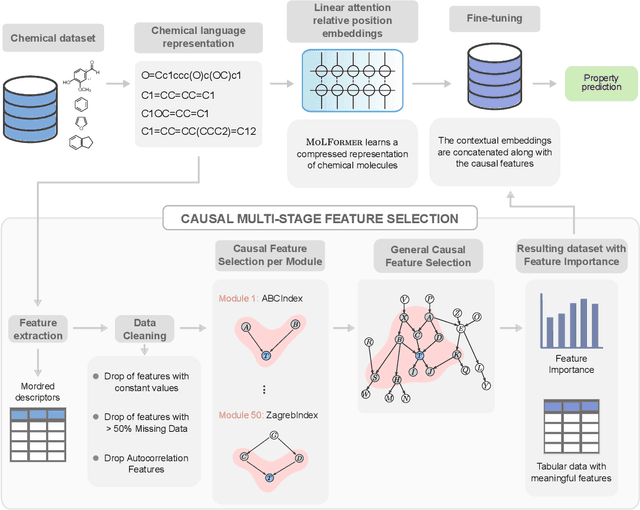

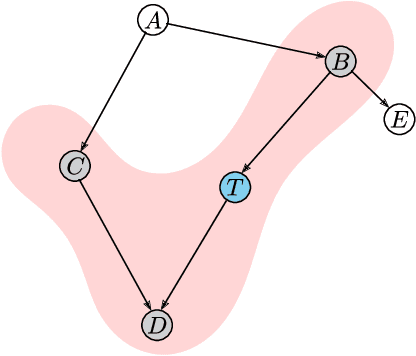
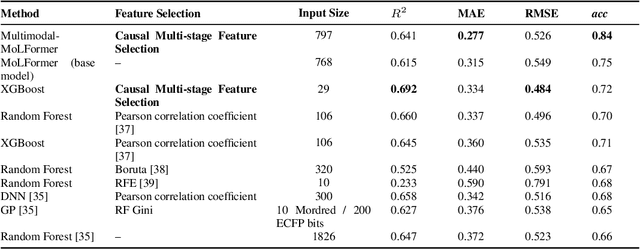
We present a novel multimodal language model approach for predicting molecular properties by combining chemical language representation with physicochemical features. Our approach, MULTIMODAL-MOLFORMER, utilizes a causal multistage feature selection method that identifies physicochemical features based on their direct causal effect on a specific target property. These causal features are then integrated with the vector space generated by molecular embeddings from MOLFORMER. In particular, we employ Mordred descriptors as physicochemical features and identify the Markov blanket of the target property, which theoretically contains the most relevant features for accurate prediction. Our results demonstrate a superior performance of our proposed approach compared to existing state-of-the-art algorithms, including the chemical language-based MOLFORMER and graph neural networks, in predicting complex tasks such as biodegradability and PFAS toxicity estimation. Moreover, we demonstrate the effectiveness of our feature selection method in reducing the dimensionality of the Mordred feature space while maintaining or improving the model's performance. Our approach opens up promising avenues for future research in molecular property prediction by harnessing the synergistic potential of both chemical language and physicochemical features, leading to enhanced performance and advancements in the field.
Human-AI Co-Creation Approach to Find Forever Chemicals Replacements
Apr 11, 2023
Generative models are a powerful tool in AI for material discovery. We are designing a software framework that supports a human-AI co-creation process to accelerate finding replacements for the ``forever chemicals''-- chemicals that enable our modern lives, but are harmful to the environment and the human health. Our approach combines AI capabilities with the domain-specific tacit knowledge of subject matter experts to accelerate the material discovery. Our co-creation process starts with the interaction between the subject matter experts and a generative model that can generate new molecule designs. In this position paper, we discuss our hypothesis that these subject matter experts can benefit from a more iterative interaction with the generative model, asking for smaller samples and ``guiding'' the exploration of the discovery space with their knowledge.
Knowledge-augmented Risk Assessment (KaRA): a hybrid-intelligence framework for supporting knowledge-intensive risk assessment of prospect candidates
Mar 09, 2023



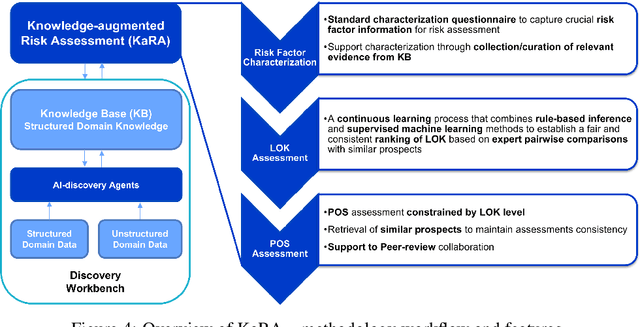
Evaluating the potential of a prospective candidate is a common task in multiple decision-making processes in different industries. We refer to a prospect as something or someone that could potentially produce positive results in a given context, e.g., an area where an oil company could find oil, a compound that, when synthesized, results in a material with required properties, and so on. In many contexts, assessing the Probability of Success (PoS) of prospects heavily depends on experts' knowledge, often leading to biased and inconsistent assessments. We have developed the framework named KARA (Knowledge-augmented Risk Assessment) to address these issues. It combines multiple AI techniques that consider SMEs (Subject Matter Experts) feedback on top of a structured domain knowledge-base to support risk assessment processes of prospect candidates in knowledge-intensive contexts.
Position Paper on Dataset Engineering to Accelerate Science
Mar 09, 2023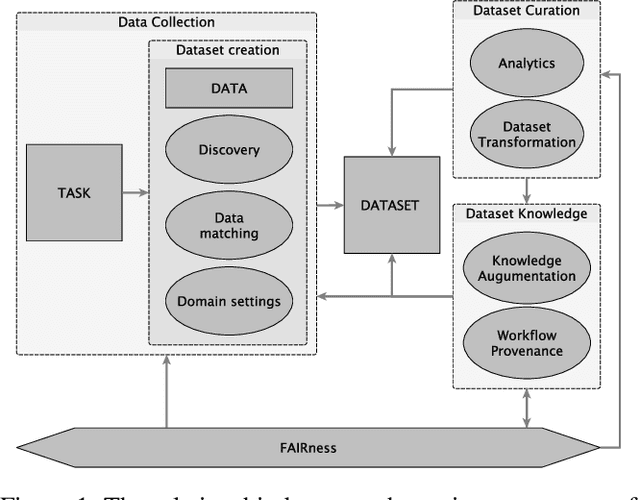
Data is a critical element in any discovery process. In the last decades, we observed exponential growth in the volume of available data and the technology to manipulate it. However, data is only practical when one can structure it for a well-defined task. For instance, we need a corpus of text broken into sentences to train a natural language machine-learning model. In this work, we will use the token \textit{dataset} to designate a structured set of data built to perform a well-defined task. Moreover, the dataset will be used in most cases as a blueprint of an entity that at any moment can be stored as a table. Specifically, in science, each area has unique forms to organize, gather and handle its datasets. We believe that datasets must be a first-class entity in any knowledge-intensive process, and all workflows should have exceptional attention to datasets' lifecycle, from their gathering to uses and evolution. We advocate that science and engineering discovery processes are extreme instances of the need for such organization on datasets, claiming for new approaches and tooling. Furthermore, these requirements are more evident when the discovery workflow uses artificial intelligence methods to empower the subject-matter expert. In this work, we discuss an approach to bringing datasets as a critical entity in the discovery process in science. We illustrate some concepts using material discovery as a use case. We chose this domain because it leverages many significant problems that can be generalized to other science fields.
Toward Human-AI Co-creation to Accelerate Material Discovery
Nov 05, 2022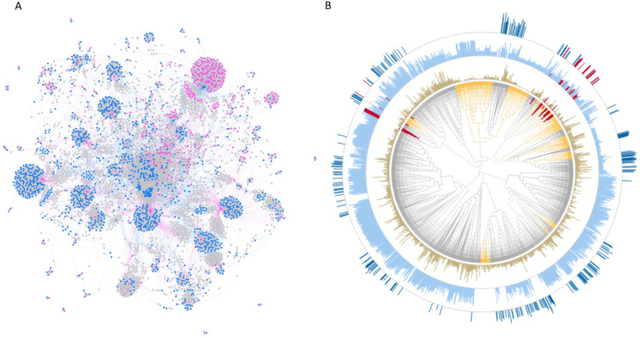
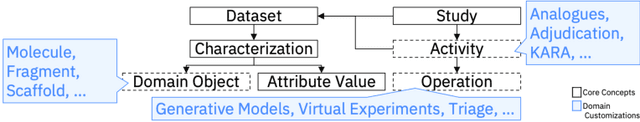
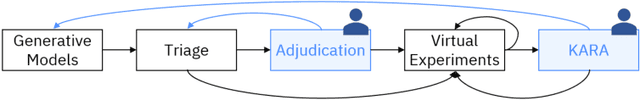
There is an increasing need in our society to achieve faster advances in Science to tackle urgent problems, such as climate changes, environmental hazards, sustainable energy systems, pandemics, among others. In certain domains like chemistry, scientific discovery carries the extra burden of assessing risks of the proposed novel solutions before moving to the experimental stage. Despite several recent advances in Machine Learning and AI to address some of these challenges, there is still a gap in technologies to support end-to-end discovery applications, integrating the myriad of available technologies into a coherent, orchestrated, yet flexible discovery process. Such applications need to handle complex knowledge management at scale, enabling knowledge consumption and production in a timely and efficient way for subject matter experts (SMEs). Furthermore, the discovery of novel functional materials strongly relies on the development of exploration strategies in the chemical space. For instance, generative models have gained attention within the scientific community due to their ability to generate enormous volumes of novel molecules across material domains. These models exhibit extreme creativity that often translates in low viability of the generated candidates. In this work, we propose a workbench framework that aims at enabling the human-AI co-creation to reduce the time until the first discovery and the opportunity costs involved. This framework relies on a knowledge base with domain and process knowledge, and user-interaction components to acquire knowledge and advise the SMEs. Currently,the framework supports four main activities: generative modeling, dataset triage, molecule adjudication, and risk assessment.
Workflow Provenance in the Lifecycle of Scientific Machine Learning
Sep 30, 2020
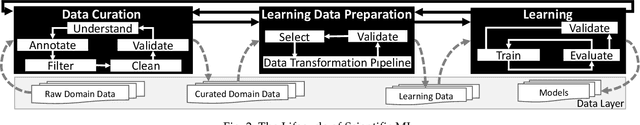
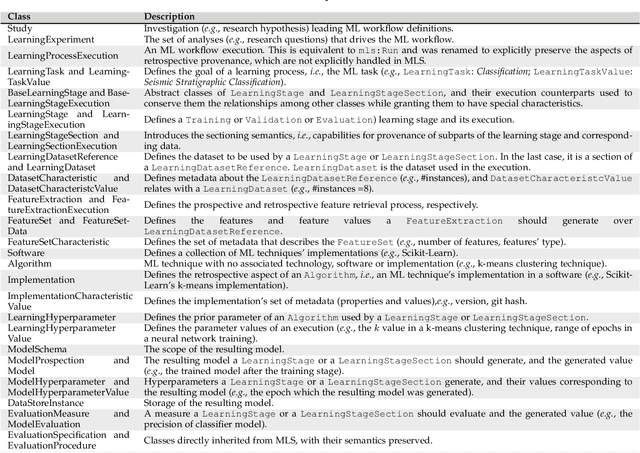
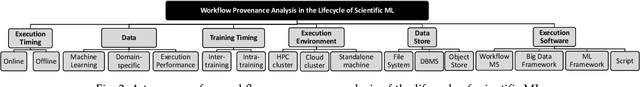
Machine Learning (ML) has already fundamentally changed several businesses. More recently, it has also been profoundly impacting the computational science and engineering domains, like geoscience, climate science, and health science. In these domains, users need to perform comprehensive data analyses combining scientific data and ML models to provide for critical requirements, such as reproducibility, model explainability, and experiment data understanding. However, scientific ML is multidisciplinary, heterogeneous, and affected by the physical constraints of the domain, making such analyses even more challenging. In this work, we leverage workflow provenance techniques to build a holistic view to support the lifecycle of scientific ML. We contribute with (i) characterization of the lifecycle and taxonomy for data analyses; (ii) design principles to build this view, with a W3C PROV compliant data representation and a reference system architecture; and (iii) lessons learned after an evaluation in an Oil & Gas case using an HPC cluster with 393 nodes and 946 GPUs. The experiments show that the principles enable queries that integrate domain semantics with ML models while keeping low overhead (<1%), high scalability, and an order of magnitude of query acceleration under certain workloads against without our representation.