 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Large Encoder-Decoder Family of Foundation Models For Chemical Language
Jul 24, 2024Large-scale pre-training methodologies for chemical language models represent a breakthrough in cheminformatics. These methods excel in tasks such as property prediction and molecule generation by learning contextualized representations of input tokens through self-supervised learning on large unlabeled corpora. Typically, this involves pre-training on unlabeled data followed by fine-tuning on specific tasks, reducing dependence on annotated datasets and broadening chemical language representation understanding. This paper introduces a large encoder-decoder chemical foundation models pre-trained on a curated dataset of 91 million SMILES samples sourced from PubChem, which is equivalent to 4 billion of molecular tokens. The proposed foundation model supports different complex tasks, including quantum property prediction, and offer flexibility with two main variants (289M and $8\times289M$). Our experiments across multiple benchmark datasets validate the capacity of the proposed model in providing state-of-the-art results for different tasks. We also provide a preliminary assessment of the compositionality of the embedding space as a prerequisite for the reasoning tasks. We demonstrate that the produced latent space is separable compared to the state-of-the-art with few-shot learning capabilities.
Formulation Graphs for Mapping Structure-Composition of Battery Electrolytes to Device Performance
Jul 27, 2023

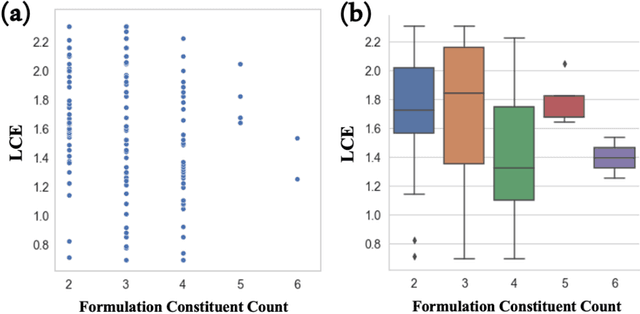
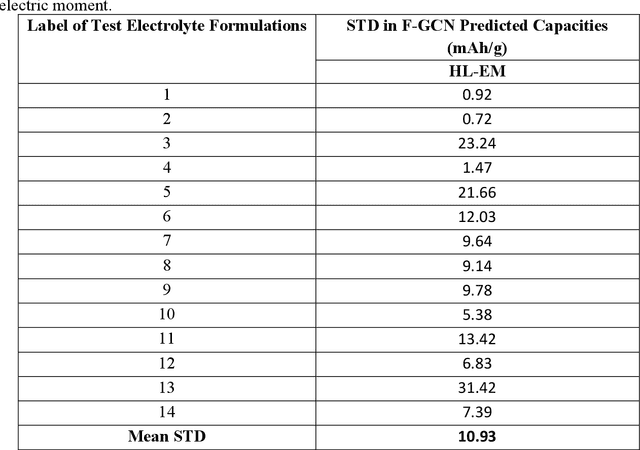
Advanced computational methods are being actively sought for addressing the challenges associated with discovery and development of new combinatorial material such as formulations. A widely adopted approach involves domain informed high-throughput screening of individual components that can be combined into a formulation. This manages to accelerate the discovery of new compounds for a target application but still leave the process of identifying the right 'formulation' from the shortlisted chemical space largely a laboratory experiment-driven process. We report a deep learning model, Formulation Graph Convolution Network (F-GCN), that can map structure-composition relationship of the individual components to the property of liquid formulation as whole. Multiple GCNs are assembled in parallel that featurize formulation constituents domain-intuitively on the fly. The resulting molecular descriptors are scaled based on respective constituent's molar percentage in the formulation, followed by formalizing into a combined descriptor that represents a complete formulation to an external learning architecture. The use case of proposed formulation learning model is demonstrated for battery electrolytes by training and testing it on two exemplary datasets representing electrolyte formulations vs battery performance -- one dataset is sourced from literature about Li/Cu half-cells, while the other is obtained by lab-experiments related to lithium-iodide full-cell chemistry. The model is shown to predict the performance metrics like Coulombic Efficiency (CE) and specific capacity of new electrolyte formulations with lowest reported errors. The best performing F-GCN model uses molecular descriptors derived from molecular graphs that are informed with HOMO-LUMO and electric moment properties of the molecules using a knowledge transfer technique.
Beyond Chemical Language: A Multimodal Approach to Enhance Molecular Property Prediction
Jun 22, 2023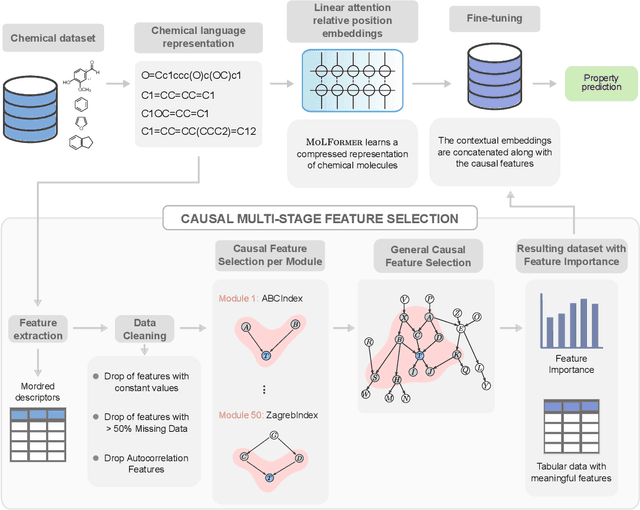

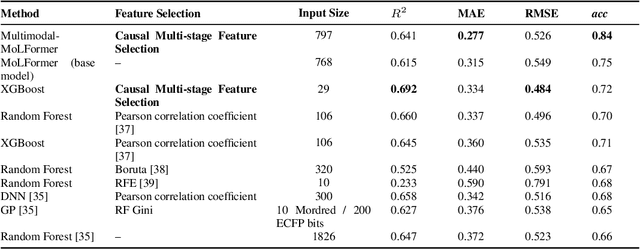
We present a novel multimodal language model approach for predicting molecular properties by combining chemical language representation with physicochemical features. Our approach, MULTIMODAL-MOLFORMER, utilizes a causal multistage feature selection method that identifies physicochemical features based on their direct causal effect on a specific target property. These causal features are then integrated with the vector space generated by molecular embeddings from MOLFORMER. In particular, we employ Mordred descriptors as physicochemical features and identify the Markov blanket of the target property, which theoretically contains the most relevant features for accurate prediction. Our results demonstrate a superior performance of our proposed approach compared to existing state-of-the-art algorithms, including the chemical language-based MOLFORMER and graph neural networks, in predicting complex tasks such as biodegradability and PFAS toxicity estimation. Moreover, we demonstrate the effectiveness of our feature selection method in reducing the dimensionality of the Mordred feature space while maintaining or improving the model's performance. Our approach opens up promising avenues for future research in molecular property prediction by harnessing the synergistic potential of both chemical language and physicochemical features, leading to enhanced performance and advancements in the field.
Human-AI Co-Creation Approach to Find Forever Chemicals Replacements
Apr 11, 2023
Generative models are a powerful tool in AI for material discovery. We are designing a software framework that supports a human-AI co-creation process to accelerate finding replacements for the ``forever chemicals''-- chemicals that enable our modern lives, but are harmful to the environment and the human health. Our approach combines AI capabilities with the domain-specific tacit knowledge of subject matter experts to accelerate the material discovery. Our co-creation process starts with the interaction between the subject matter experts and a generative model that can generate new molecule designs. In this position paper, we discuss our hypothesis that these subject matter experts can benefit from a more iterative interaction with the generative model, asking for smaller samples and ``guiding'' the exploration of the discovery space with their knowledge.
Domain-agnostic and Multi-level Evaluation of Generative Models
Jan 20, 2023



While the capabilities of generative models heavily improved in different domains (images, text, graphs, molecules, etc.), their evaluation metrics largely remain based on simplified quantities or manual inspection with limited practicality. To this end, we propose a framework for Multi-level Performance Evaluation of Generative mOdels (MPEGO), which could be employed across different domains. MPEGO aims to quantify generation performance hierarchically, starting from a sub-feature-based low-level evaluation to a global features-based high-level evaluation. MPEGO offers great customizability as the employed features are entirely user-driven and can thus be highly domain/problem-specific while being arbitrarily complex (e.g., outcomes of experimental procedures). We validate MPEGO using multiple generative models across several datasets from the material discovery domain. An ablation study is conducted to study the plausibility of intermediate steps in MPEGO. Results demonstrate that MPEGO provides a flexible, user-driven, and multi-level evaluation framework, with practical insights on the generation quality. The framework, source code, and experiments will be available at https://github.com/GT4SD/mpego.
Toward Human-AI Co-creation to Accelerate Material Discovery
Nov 05, 2022
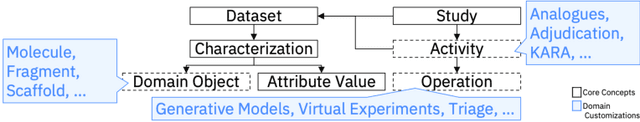
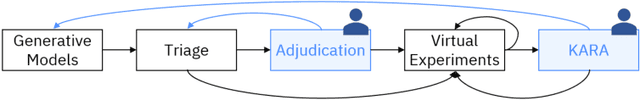
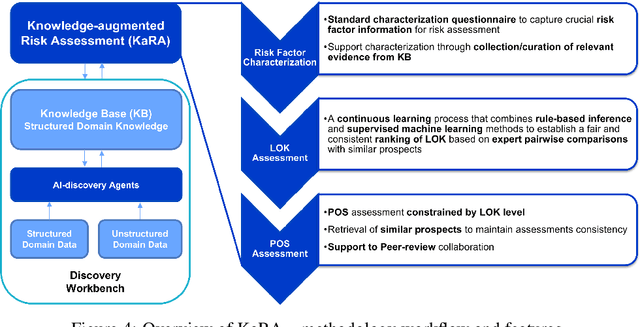
There is an increasing need in our society to achieve faster advances in Science to tackle urgent problems, such as climate changes, environmental hazards, sustainable energy systems, pandemics, among others. In certain domains like chemistry, scientific discovery carries the extra burden of assessing risks of the proposed novel solutions before moving to the experimental stage. Despite several recent advances in Machine Learning and AI to address some of these challenges, there is still a gap in technologies to support end-to-end discovery applications, integrating the myriad of available technologies into a coherent, orchestrated, yet flexible discovery process. Such applications need to handle complex knowledge management at scale, enabling knowledge consumption and production in a timely and efficient way for subject matter experts (SMEs). Furthermore, the discovery of novel functional materials strongly relies on the development of exploration strategies in the chemical space. For instance, generative models have gained attention within the scientific community due to their ability to generate enormous volumes of novel molecules across material domains. These models exhibit extreme creativity that often translates in low viability of the generated candidates. In this work, we propose a workbench framework that aims at enabling the human-AI co-creation to reduce the time until the first discovery and the opportunity costs involved. This framework relies on a knowledge base with domain and process knowledge, and user-interaction components to acquire knowledge and advise the SMEs. Currently,the framework supports four main activities: generative modeling, dataset triage, molecule adjudication, and risk assessment.
Molecular Inverse-Design Platform for Material Industries
Apr 27, 2020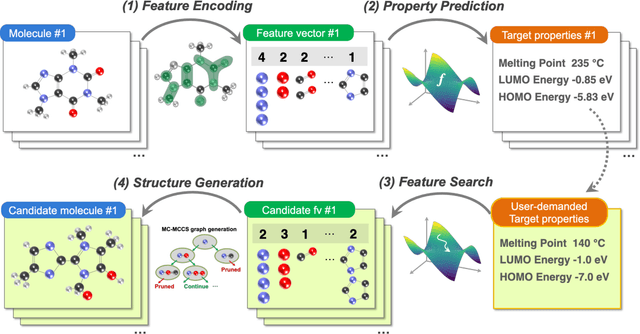
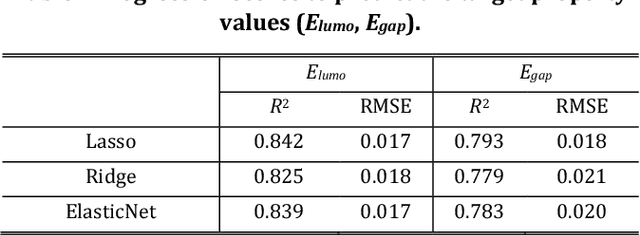
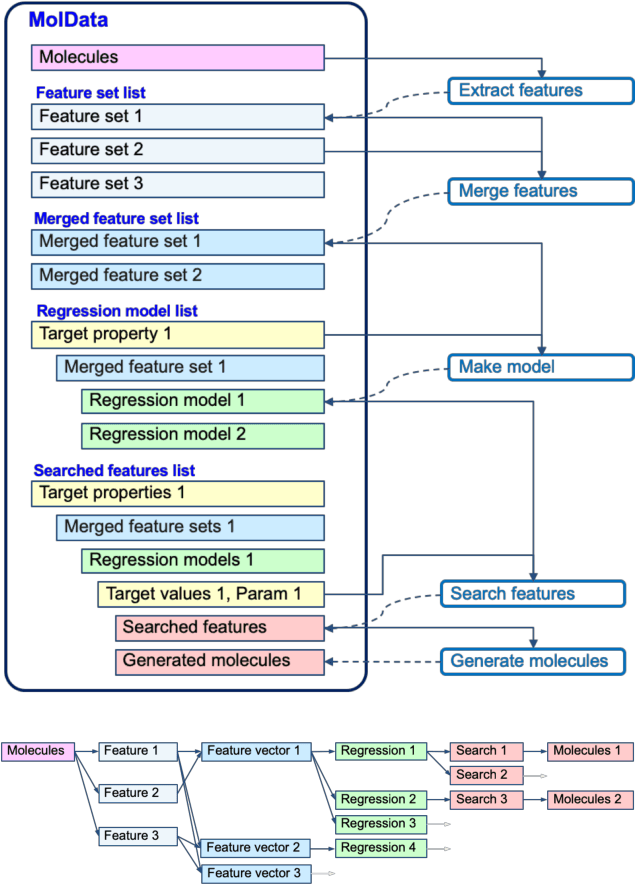
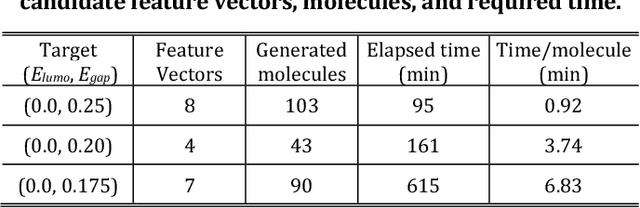
The discovery of new materials has been the essential force which brings a discontinuous improvement to industrial products' performance. However, the extra-vast combinatorial design space of material structures exceeds human experts' capability to explore all, thereby hampering material development. In this paper, we present a material industry-oriented web platform of an AI-driven molecular inverse-design system, which automatically designs brand new molecular structures rapidly and diversely. Different from existing inverse-design solutions, in this system, the combination of substructure-based feature encoding and molecular graph generation algorithms allows a user to gain high-speed, interpretable, and customizable design process. Also, a hierarchical data structure and user-oriented UI provide a flexible and intuitive workflow. The system is deployed on IBM's and our client's cloud servers and has been used by 5 partner companies. To illustrate actual industrial use cases, we exhibit inverse-design of sugar and dye molecules, that were carried out by experimental chemists in those client companies. Compared to general human chemist's standard performance, the molecular design speed was accelerated more than 10 times, and greatly increased variety was observed in the inverse-designed molecules without loss of chemical realism.
Data Infrastructure and Approaches for Ontology-Based Drug Repurposing
Jul 12, 2018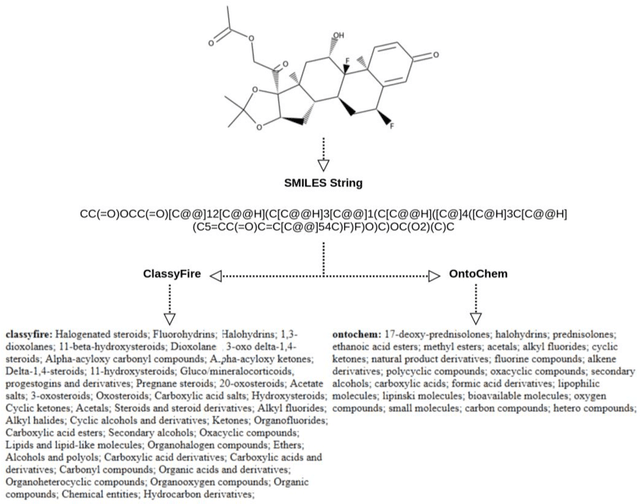

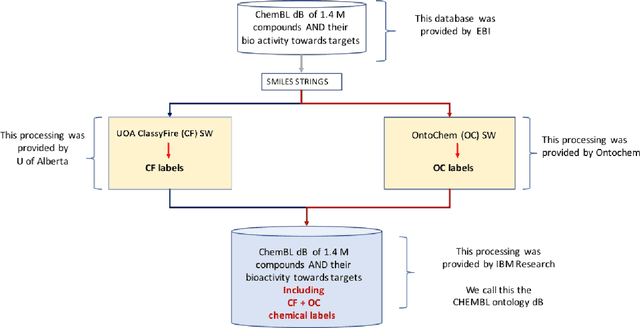

We report development of a data infrastructure for drug repurposing that takes advantage of two currently available chemical ontologies. The data infrastructure includes a database of compound- target associations augmented with molecular ontological labels. It also contains two computational tools for prediction of new associations. We describe two drug-repurposing systems: one, Nascent Ontological Information Retrieval for Drug Repurposing (NOIR-DR), based on an information retrieval strategy, and another, based on non-negative matrix factorization together with compound similarity, that was inspired by recommender systems. We report the performance of both tools on a drug-repurposing task.