 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImproving Electrolyte Performance for Target Cathode Loading Using Interpretable Data-Driven Approach
Sep 03, 2024
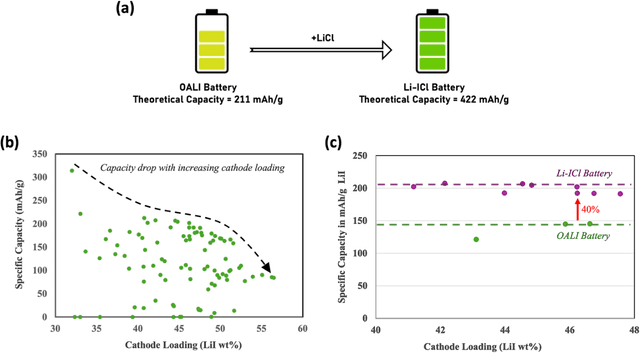
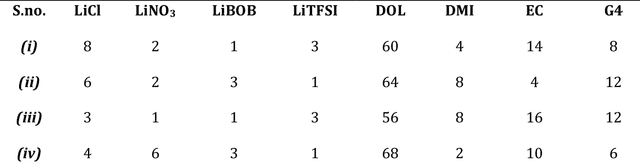

Higher loading of active electrode materials is desired in batteries, especially those based on conversion reactions, for enhanced energy density and cost efficiency. However, increasing active material loading in electrodes can cause significant performance depreciation due to internal resistance, shuttling, and parasitic side reactions, which can be alleviated to a certain extent by a compatible design of electrolytes. In this work, a data-driven approach is leveraged to find a high-performing electrolyte formulation for a novel interhalogen battery custom to the target cathode loading. An electrolyte design consisting of 4 solvents and 4 salts is experimentally devised for a novel interhalogen battery based on a multi-electron redox reaction. The experimental dataset with variable electrolyte compositions and active cathode loading, is used to train a graph-based deep learning model mapping changing variables in the battery's material design to its specific capacity. The trained model is used to further optimize the electrolyte formulation compositions for enhancing the battery capacity at a target cathode loading by a two-fold approach: large-scale screening and interpreting electrolyte design principles for different cathode loadings. The data-driven approach is demonstrated to bring about an additional 20% increment in the specific capacity of the battery over capacities obtained from the experimental optimization.
Improving Performance Prediction of Electrolyte Formulations with Transformer-based Molecular Representation Model
Jun 28, 2024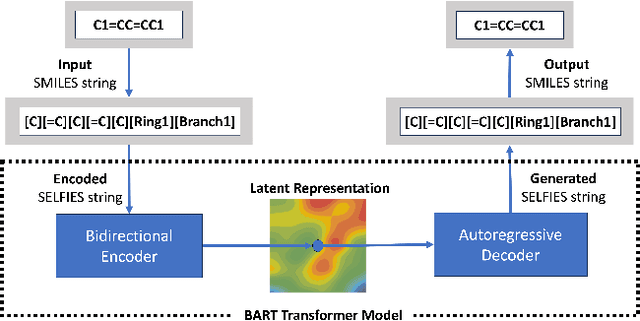

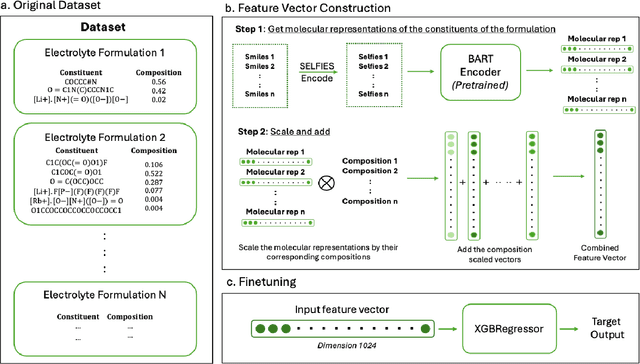

Development of efficient and high-performing electrolytes is crucial for advancing energy storage technologies, particularly in batteries. Predicting the performance of battery electrolytes rely on complex interactions between the individual constituents. Consequently, a strategy that adeptly captures these relationships and forms a robust representation of the formulation is essential for integrating with machine learning models to predict properties accurately. In this paper, we introduce a novel approach leveraging a transformer-based molecular representation model to effectively and efficiently capture the representation of electrolyte formulations. The performance of the proposed approach is evaluated on two battery property prediction tasks and the results show superior performance compared to the state-of-the-art methods.
Formulation Graphs for Mapping Structure-Composition of Battery Electrolytes to Device Performance
Jul 27, 2023

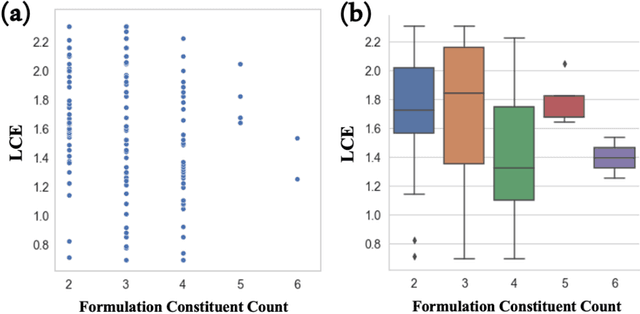
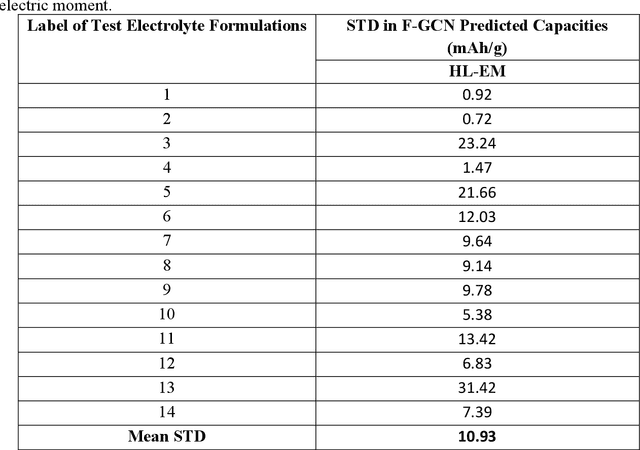
Advanced computational methods are being actively sought for addressing the challenges associated with discovery and development of new combinatorial material such as formulations. A widely adopted approach involves domain informed high-throughput screening of individual components that can be combined into a formulation. This manages to accelerate the discovery of new compounds for a target application but still leave the process of identifying the right 'formulation' from the shortlisted chemical space largely a laboratory experiment-driven process. We report a deep learning model, Formulation Graph Convolution Network (F-GCN), that can map structure-composition relationship of the individual components to the property of liquid formulation as whole. Multiple GCNs are assembled in parallel that featurize formulation constituents domain-intuitively on the fly. The resulting molecular descriptors are scaled based on respective constituent's molar percentage in the formulation, followed by formalizing into a combined descriptor that represents a complete formulation to an external learning architecture. The use case of proposed formulation learning model is demonstrated for battery electrolytes by training and testing it on two exemplary datasets representing electrolyte formulations vs battery performance -- one dataset is sourced from literature about Li/Cu half-cells, while the other is obtained by lab-experiments related to lithium-iodide full-cell chemistry. The model is shown to predict the performance metrics like Coulombic Efficiency (CE) and specific capacity of new electrolyte formulations with lowest reported errors. The best performing F-GCN model uses molecular descriptors derived from molecular graphs that are informed with HOMO-LUMO and electric moment properties of the molecules using a knowledge transfer technique.