 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProtoMedAgent: Multimodal Clinical Interpretability via Privacy-Aware Agentic Workflows
May 13, 2026While interpretable prototype networks offer compelling case-based reasoning for clinical diagnostics, their raw continuous outputs lack the semantic structure required for medical documentation. Bridging this gap via standard Retrieval-Augmented Generation (RAG) routinely triggers ``retrieval sycophancy,'' where Large Language Models (LLMs) hallucinate post-hoc rationalizations to align with visual predictions. We introduce ProtoMedAgent, a framework that formalizes multimodal clinical reporting as an iterative, zero-gradient test-time optimization problem over a strict neuro-symbolic bottleneck. Operating on a frozen prototype backbone, we distill latent visual and tabular features into a discrete semantic memory. Online generation is strictly constrained by exact set-theoretic differentials and a reflective Scribe-Critic loop, mathematically precluding unsupported narrative claims. To safely bound data disclosure, we introduce a semantic privacy gate governed by $k$-anonymity and $\ell$-diversity. Evaluated on a 4,160-patient clinical cohort, ProtoMedAgent achieves 91.2\% Comparison Set Faithfulness where it fundamentally outperforms standard RAG (46.2\%). ProtoMedAgent additionally leverages a binding $\ell$-diversity phase transition to systematically reduce artifact-level membership inference risks by an absolute 9.8\%.
A Large Encoder-Decoder Family of Foundation Models For Chemical Language
Jul 24, 2024Large-scale pre-training methodologies for chemical language models represent a breakthrough in cheminformatics. These methods excel in tasks such as property prediction and molecule generation by learning contextualized representations of input tokens through self-supervised learning on large unlabeled corpora. Typically, this involves pre-training on unlabeled data followed by fine-tuning on specific tasks, reducing dependence on annotated datasets and broadening chemical language representation understanding. This paper introduces a large encoder-decoder chemical foundation models pre-trained on a curated dataset of 91 million SMILES samples sourced from PubChem, which is equivalent to 4 billion of molecular tokens. The proposed foundation model supports different complex tasks, including quantum property prediction, and offer flexibility with two main variants (289M and $8\times289M$). Our experiments across multiple benchmark datasets validate the capacity of the proposed model in providing state-of-the-art results for different tasks. We also provide a preliminary assessment of the compositionality of the embedding space as a prerequisite for the reasoning tasks. We demonstrate that the produced latent space is separable compared to the state-of-the-art with few-shot learning capabilities.
Improving Molecular Properties Prediction Through Latent Space Fusion
Oct 20, 2023Pre-trained Language Models have emerged as promising tools for predicting molecular properties, yet their development is in its early stages, necessitating further research to enhance their efficacy and address challenges such as generalization and sample efficiency. In this paper, we present a multi-view approach that combines latent spaces derived from state-of-the-art chemical models. Our approach relies on two pivotal elements: the embeddings derived from MHG-GNN, which represent molecular structures as graphs, and MoLFormer embeddings rooted in chemical language. The attention mechanism of MoLFormer is able to identify relations between two atoms even when their distance is far apart, while the GNN of MHG-GNN can more precisely capture relations among multiple atoms closely located. In this work, we demonstrate the superior performance of our proposed multi-view approach compared to existing state-of-the-art methods, including MoLFormer-XL, which was trained on 1.1 billion molecules, particularly in intricate tasks such as predicting clinical trial drug toxicity and inhibiting HIV replication. We assessed our approach using six benchmark datasets from MoleculeNet, where it outperformed competitors in five of them. Our study highlights the potential of latent space fusion and feature integration for advancing molecular property prediction. In this work, we use small versions of MHG-GNN and MoLFormer, which opens up an opportunity for further improvement when our approach uses a larger-scale dataset.
Beyond Chemical Language: A Multimodal Approach to Enhance Molecular Property Prediction
Jun 22, 2023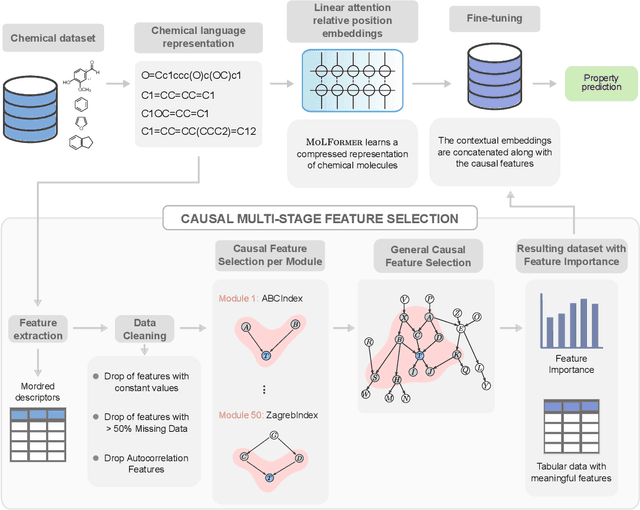

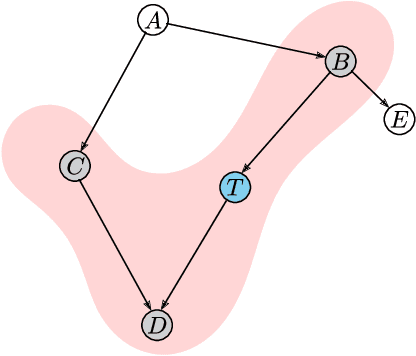
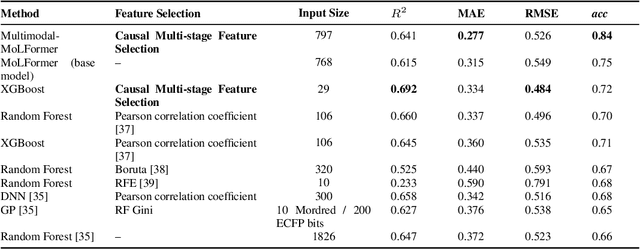
We present a novel multimodal language model approach for predicting molecular properties by combining chemical language representation with physicochemical features. Our approach, MULTIMODAL-MOLFORMER, utilizes a causal multistage feature selection method that identifies physicochemical features based on their direct causal effect on a specific target property. These causal features are then integrated with the vector space generated by molecular embeddings from MOLFORMER. In particular, we employ Mordred descriptors as physicochemical features and identify the Markov blanket of the target property, which theoretically contains the most relevant features for accurate prediction. Our results demonstrate a superior performance of our proposed approach compared to existing state-of-the-art algorithms, including the chemical language-based MOLFORMER and graph neural networks, in predicting complex tasks such as biodegradability and PFAS toxicity estimation. Moreover, we demonstrate the effectiveness of our feature selection method in reducing the dimensionality of the Mordred feature space while maintaining or improving the model's performance. Our approach opens up promising avenues for future research in molecular property prediction by harnessing the synergistic potential of both chemical language and physicochemical features, leading to enhanced performance and advancements in the field.
Position Paper on Dataset Engineering to Accelerate Science
Mar 09, 2023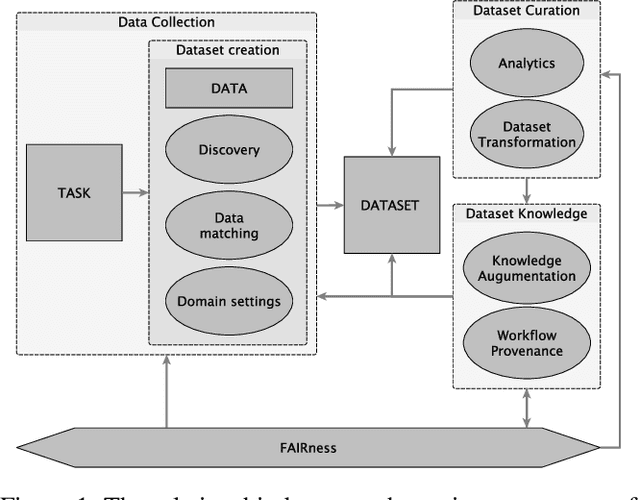
Data is a critical element in any discovery process. In the last decades, we observed exponential growth in the volume of available data and the technology to manipulate it. However, data is only practical when one can structure it for a well-defined task. For instance, we need a corpus of text broken into sentences to train a natural language machine-learning model. In this work, we will use the token \textit{dataset} to designate a structured set of data built to perform a well-defined task. Moreover, the dataset will be used in most cases as a blueprint of an entity that at any moment can be stored as a table. Specifically, in science, each area has unique forms to organize, gather and handle its datasets. We believe that datasets must be a first-class entity in any knowledge-intensive process, and all workflows should have exceptional attention to datasets' lifecycle, from their gathering to uses and evolution. We advocate that science and engineering discovery processes are extreme instances of the need for such organization on datasets, claiming for new approaches and tooling. Furthermore, these requirements are more evident when the discovery workflow uses artificial intelligence methods to empower the subject-matter expert. In this work, we discuss an approach to bringing datasets as a critical entity in the discovery process in science. We illustrate some concepts using material discovery as a use case. We chose this domain because it leverages many significant problems that can be generalized to other science fields.
An Interpretable Deep Semantic Segmentation Method for Earth Observation
Oct 23, 2022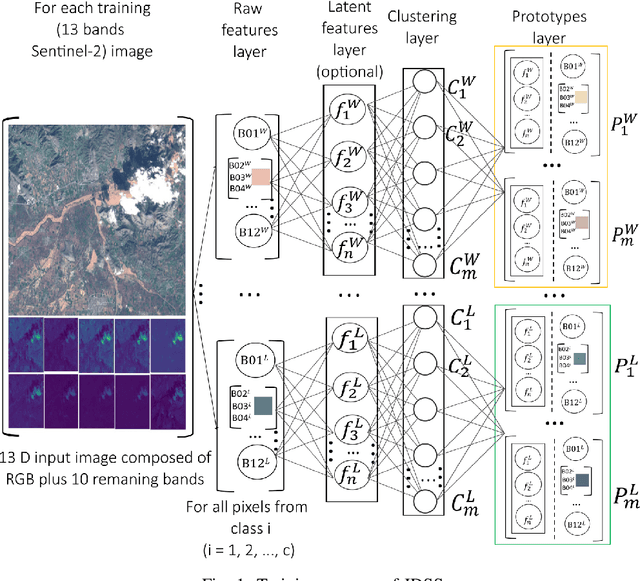
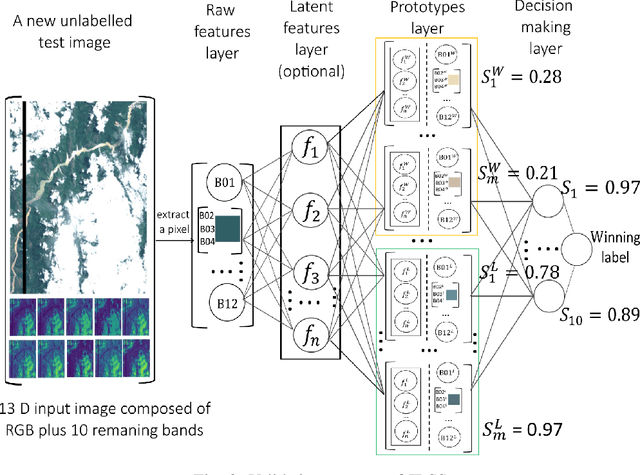
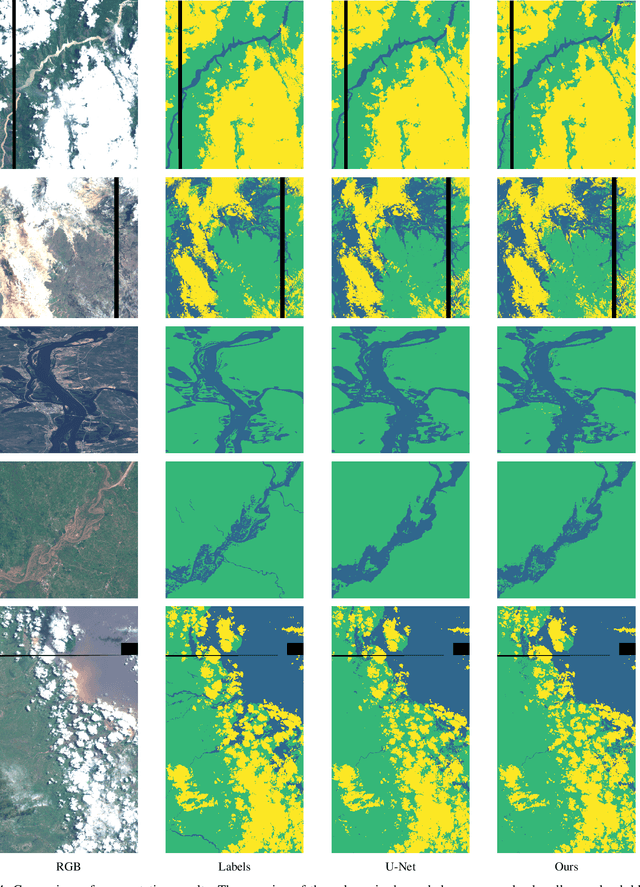
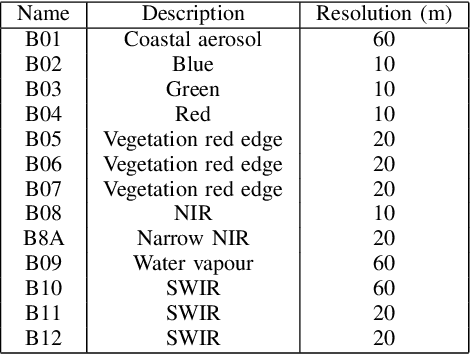
Earth observation is fundamental for a range of human activities including flood response as it offers vital information to decision makers. Semantic segmentation plays a key role in mapping the raw hyper-spectral data coming from the satellites into a human understandable form assigning class labels to each pixel. In this paper, we introduce a prototype-based interpretable deep semantic segmentation (IDSS) method, which is highly accurate as well as interpretable. Its parameters are in orders of magnitude less than the number of parameters used by deep networks such as U-Net and are clearly interpretable by humans. The proposed here IDSS offers a transparent structure that allows users to inspect and audit the algorithm's decision. Results have demonstrated that IDSS could surpass other algorithms, including U-Net, in terms of IoU (Intersection over Union) total water and Recall total water. We used WorldFloods data set for our experiments and plan to use the semantic segmentation results combined with masks for permanent water to detect flood events.
Towards Deep Machine Reasoning: a Prototype-based Deep Neural Network with Decision Tree Inference
Feb 02, 2020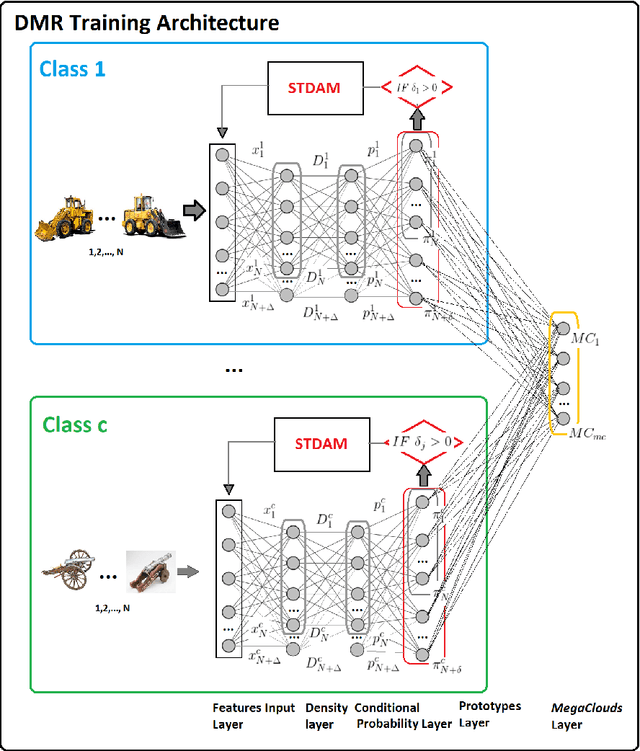


In this paper we introduce the DMR -- a prototype-based method and network architecture for deep learning which is using a decision tree (DT)-based inference and synthetic data to balance the classes. It builds upon the recently introduced xDNN method addressing more complex multi-class problems, specifically when classes are highly imbalanced. DMR moves away from a direct decision based on all classes towards a layered DT of pair-wise class comparisons. In addition, it forces the prototypes to be balanced between classes regardless of possible class imbalances of the training data. It has two novel mechanisms, namely i) using a DT to determine the winning class label, and ii) balancing the classes by synthesizing data around the prototypes determined from the available training data. As a result, we improved significantly the performance of the resulting fully explainable DNN as evidenced by the best reported result on the well know benchmark problem Caltech-101 surpassing our own recently published "world record". Furthermore, we also achieved another "world record" for another very hard benchmark problem, namely Caltech-256 as well as surpassed the results of other approaches on Faces-1999 problem. In summary, we propose a new approach specifically advantageous for imbalanced multi-class problems that achieved two world records on well known hard benchmark problems and the best result on another problem in terms of accuracy. Moreover, DMR offers full explainability, does not require GPUs and can continue to learn from new data by adding new prototypes preserving the previous ones but not requiring full retraining.
Towards Explainable Deep Neural Networks (xDNN)
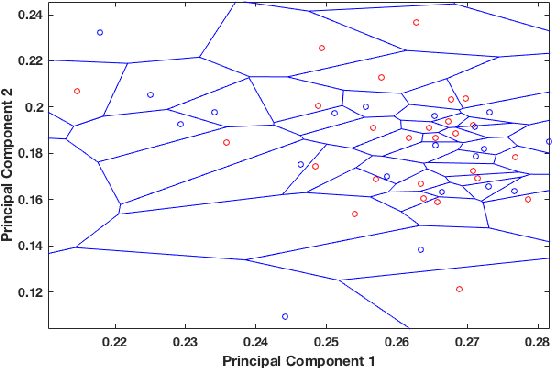
Dec 05, 2019
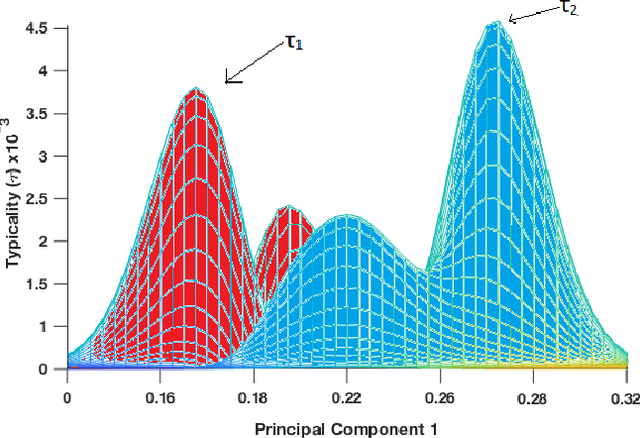
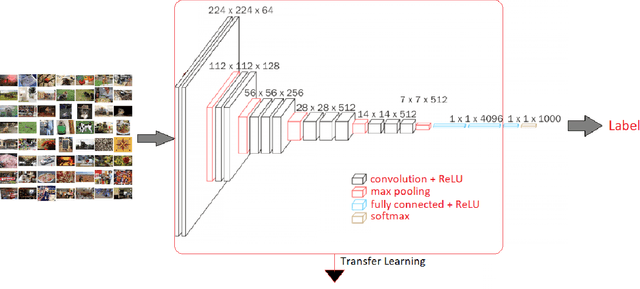

In this paper, we propose an elegant solution that is directly addressing the bottlenecks of the traditional deep learning approaches and offers a clearly explainable internal architecture that can outperform the existing methods, requires very little computational resources (no need for GPUs) and short training times (in the order of seconds). The proposed approach, xDNN is using prototypes. Prototypes are actual training data samples (images), which are local peaks of the empirical data distribution called typicality as well as of the data density. This generative model is identified in a closed form and equates to the pdf but is derived automatically and entirely from the training data with no user- or problem-specific thresholds, parameters or intervention. The proposed xDNN offers a new deep learning architecture that combines reasoning and learning in a synergy. It is non-iterative and non-parametric, which explains its efficiency in terms of time and computational resources. From the user perspective, the proposed approach is clearly understandable to human users. We tested it on some well-known benchmark data sets such as iRoads and Caltech-256. xDNN outperforms the other methods including deep learning in terms of accuracy, time to train and offers a clearly explainable classifier. In fact, the result on the very hard Caltech-256 problem (which has 257 classes) represents a world record.
Novelty Detection and Learning from Extremely Weak Supervision
Nov 01, 2019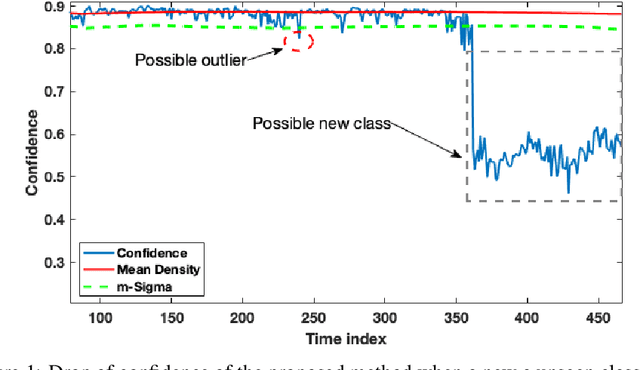
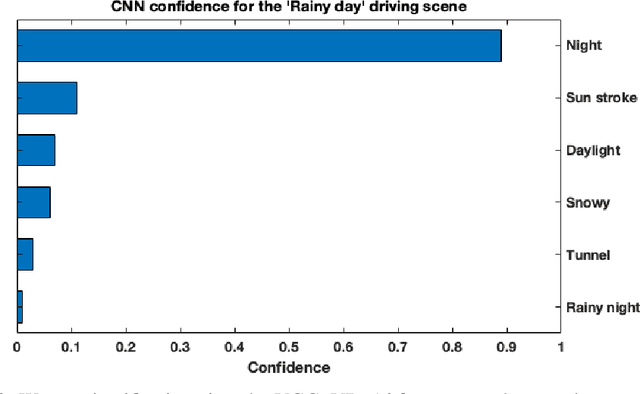
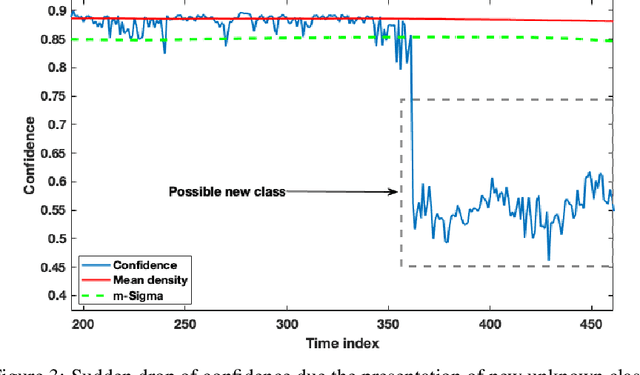

In this paper we offer a method and algorithm, which make possible fully autonomous (unsupervised) detection of new classes, and learning following a very parsimonious training priming (few labeled data samples only). Moreover, new unknown classes may appear at a later stage and the proposed xClass method and algorithm are able to successfully discover this and learn from the data autonomously. Furthermore, the features (inputs to the classifier) are automatically sub-selected by the algorithm based on the accumulated data density per feature per class. As a result, a highly efficient, lean, human-understandable, autonomously self-learning model (which only needs an extremely parsimonious priming) emerges from the data. To validate our proposal we tested it on two challenging problems, including imbalanced Caltech-101 data set and iRoads dataset. Not only we achieved higher precision, but, more significantly, we only used a single class beforehand, while other methods used all the available classes) and we generated interpretable models with smaller number of features used, through extremely weak and weak supervision.
Fair-by-design explainable models for prediction of recidivism
Sep 18, 2019
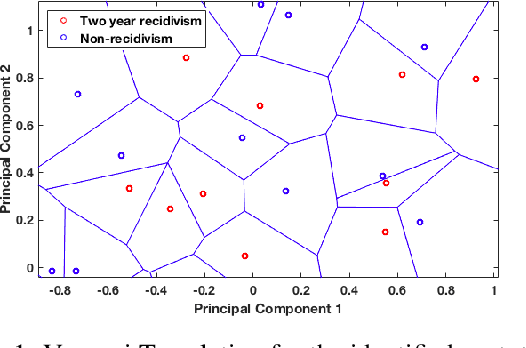

Recidivism prediction provides decision makers with an assessment of the likelihood that a criminal defendant will reoffend that can be used in pre-trial decision-making. It can also be used for prediction of locations where crimes most occur, profiles that are more likely to commit violent crimes. While such instruments are gaining increasing popularity, their use is controversial as they may present potential discriminatory bias in the risk assessment. In this paper we propose a new fair-by-design approach to predict recidivism. It is prototype-based, learns locally and extracts empirically the data distribution. The results show that the proposed method is able to reduce the bias and provide human interpretable rules to assist specialists in the explanation of the given results.