 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGT4SD: Generative Toolkit for Scientific Discovery
Jul 08, 2022
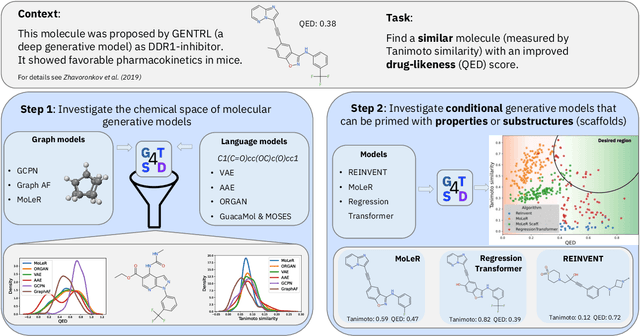
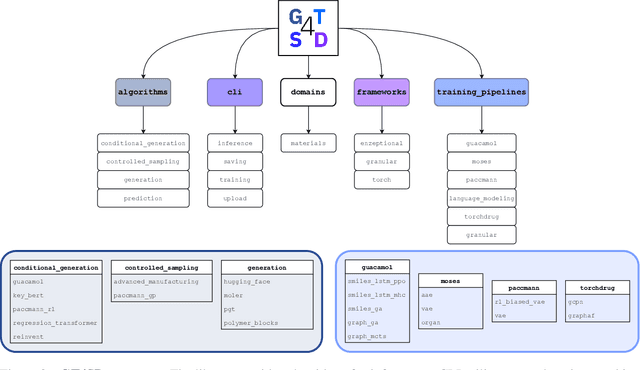
With the growing availability of data within various scientific domains, generative models hold enormous potential to accelerate scientific discovery at every step of the scientific method. Perhaps their most valuable application lies in the speeding up of what has traditionally been the slowest and most challenging step of coming up with a hypothesis. Powerful representations are now being learned from large volumes of data to generate novel hypotheses, which is making a big impact on scientific discovery applications ranging from material design to drug discovery. The GT4SD (https://github.com/GT4SD/gt4sd-core) is an extensible open-source library that enables scientists, developers and researchers to train and use state-of-the-art generative models for hypothesis generation in scientific discovery. GT4SD supports a variety of uses of generative models across material science and drug discovery, including molecule discovery and design based on properties related to target proteins, omic profiles, scaffold distances, binding energies and more.
Pre-training Protein Language Models with Label-Agnostic Binding Pairs Enhances Performance in Downstream Tasks
Dec 05, 2020



Less than 1% of protein sequences are structurally and functionally annotated. Natural Language Processing (NLP) community has recently embraced self-supervised learning as a powerful approach to learn representations from unlabeled text, in large part due to the attention-based context-aware Transformer models. In this work we present a modification to the RoBERTa model by inputting during pre-training a mixture of binding and non-binding protein sequences (from STRING database). However, the sequence pairs have no label to indicate their binding status, as the model relies solely on Masked Language Modeling (MLM) objective during pre-training. After fine-tuning, such approach surpasses models trained on single protein sequences for protein-protein binding prediction, TCR-epitope binding prediction, cellular-localization and remote homology classification tasks. We suggest that the Transformer's attention mechanism contributes to protein binding site discovery. Furthermore, we compress protein sequences by 64% with the Byte Pair Encoding (BPE) vocabulary consisting of 10K subwords, each around 3-4 amino acids long. Finally, to expand the model input space to even larger proteins and multi-protein assemblies, we pre-train Longformer models that support 2,048 tokens. Further work in token-level classification for secondary structure prediction is needed. Code available at: https://github.com/PaccMann/paccmann_proteomics
PaccMann$^{RL}$ on SARS-CoV-2: Designing antiviral candidates with conditional generative models
May 31, 2020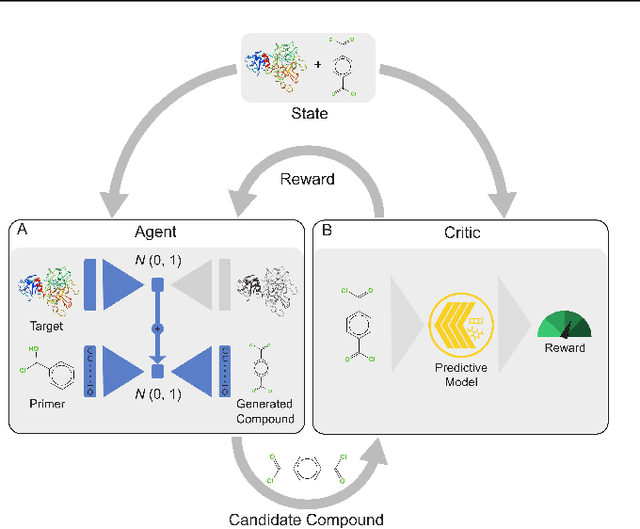

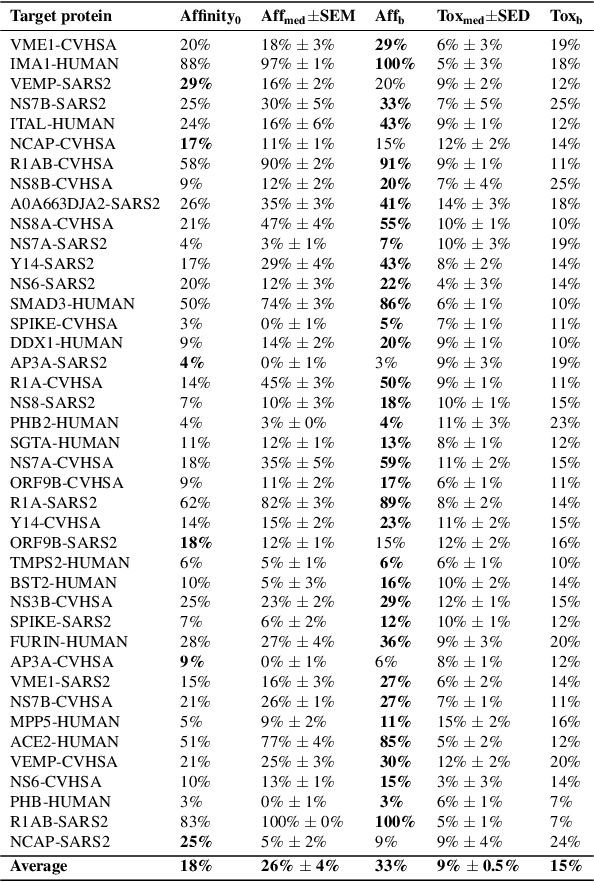
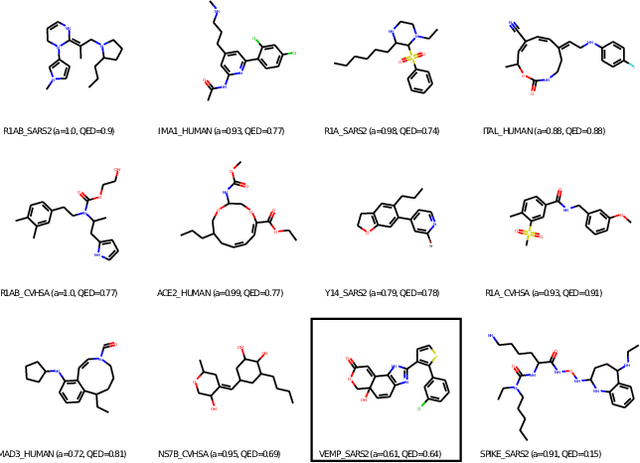
With the fast development of COVID-19 into a global pandemic, scientists around the globe are desperately searching for effective antiviral therapeutic agents. Bridging systems biology and drug discovery, we propose a deep learning framework for conditional de novo design of antiviral candidate drugs tailored against given protein targets. First, we train a multimodal ligand--protein binding affinity model on predicting affinities of antiviral compounds to target proteins and couple this model with pharmacological toxicity predictors. Exploiting this multi-objective as a reward function of a conditional molecular generator (consisting of two VAEs), we showcase a framework that navigates the chemical space toward regions with more antiviral molecules. Specifically, we explore a challenging setting of generating ligands against unseen protein targets by performing a leave-one-out-cross-validation on 41 SARS-CoV-2-related target proteins. Using deep RL, it is demonstrated that in 35 out of 41 cases, the generation is biased towards sampling more binding ligands, with an average increase of 83% comparing to an unbiased VAE. We present a case-study on a potential Envelope-protein inhibitor and perform a synthetic accessibility assessment of the best generated molecules is performed that resembles a viable roadmap towards a rapid in-vitro evaluation of potential SARS-CoV-2 inhibitors.
PIMKL: Pathway Induced Multiple Kernel Learning
Jul 05, 2018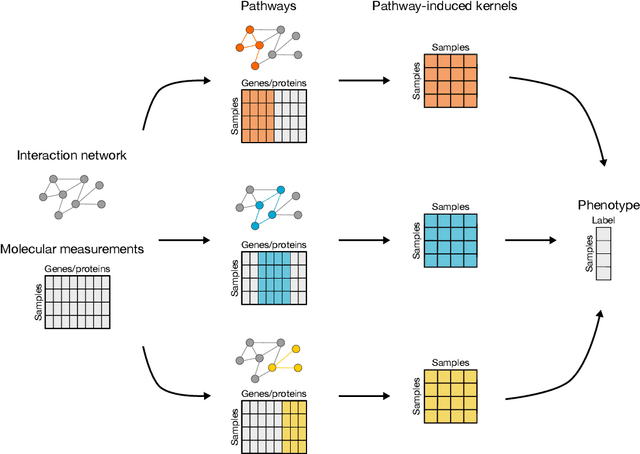
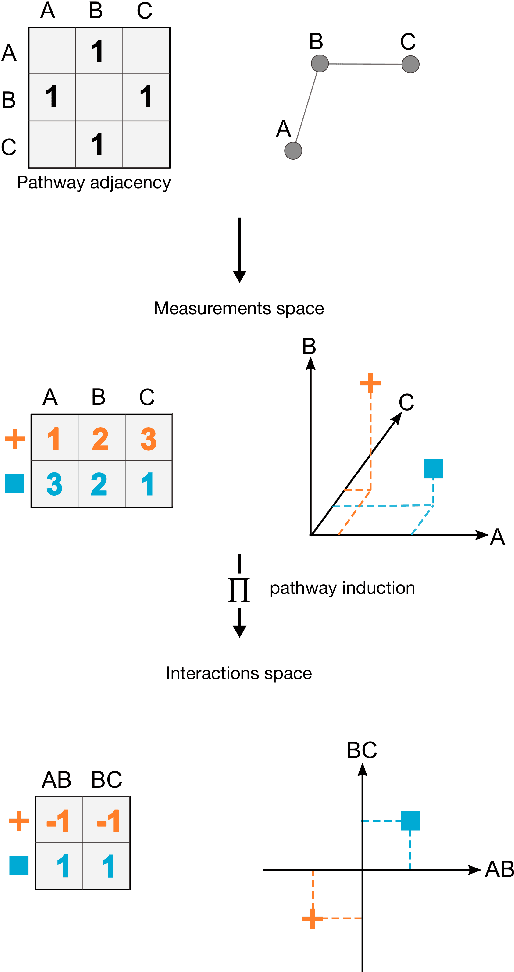
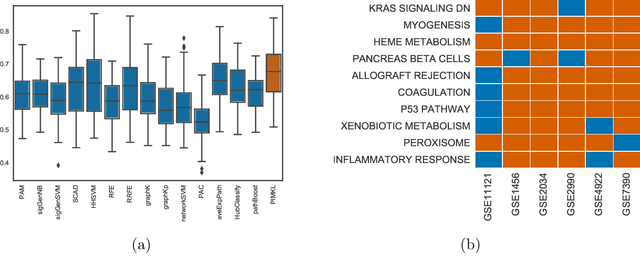
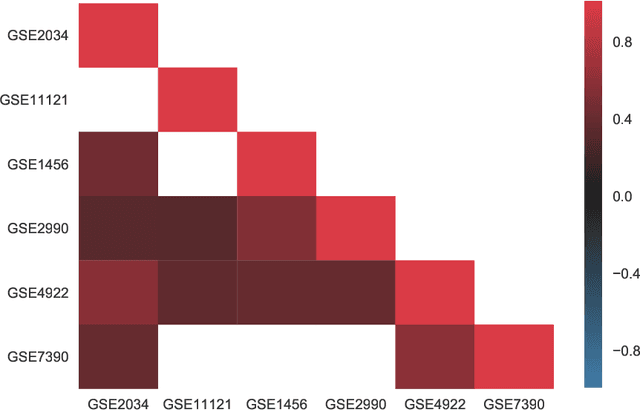
Reliable identification of molecular biomarkers is essential for accurate patient stratification. While state-of-the-art machine learning approaches for sample classification continue to push boundaries in terms of performance, most of these methods are not able to integrate different data types and lack generalization power, limiting their application in a clinical setting. Furthermore, many methods behave as black boxes, and we have very little understanding about the mechanisms that lead to the prediction. While opaqueness concerning machine behaviour might not be a problem in deterministic domains, in health care, providing explanations about the molecular factors and phenotypes that are driving the classification is crucial to build trust in the performance of the predictive system. We propose Pathway Induced Multiple Kernel Learning (PIMKL), a novel methodology to reliably classify samples that can also help gain insights into the molecular mechanisms that underlie the classification. PIMKL exploits prior knowledge in the form of a molecular interaction network and annotated gene sets, by optimizing a mixture of pathway-induced kernels using a Multiple Kernel Learning (MKL) algorithm, an approach that has demonstrated excellent performance in different machine learning applications. After optimizing the combination of kernels for prediction of a specific phenotype, the model provides a stable molecular signature that can be interpreted in the light of the ingested prior knowledge and that can be used in transfer learning tasks.