 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDockGame: Cooperative Games for Multimeric Rigid Protein Docking
Oct 09, 2023Protein interactions and assembly formation are fundamental to most biological processes. Predicting the assembly structure from constituent proteins -- referred to as the protein docking task -- is thus a crucial step in protein design applications. Most traditional and deep learning methods for docking have focused mainly on binary docking, following either a search-based, regression-based, or generative modeling paradigm. In this paper, we focus on the less-studied multimeric (i.e., two or more proteins) docking problem. We introduce DockGame, a novel game-theoretic framework for docking -- we view protein docking as a cooperative game between proteins, where the final assembly structure(s) constitute stable equilibria w.r.t. the underlying game potential. Since we do not have access to the true potential, we consider two approaches - i) learning a surrogate game potential guided by physics-based energy functions and computing equilibria by simultaneous gradient updates, and ii) sampling from the Gibbs distribution of the true potential by learning a diffusion generative model over the action spaces (rotations and translations) of all proteins. Empirically, on the Docking Benchmark 5.5 (DB5.5) dataset, DockGame has much faster runtimes than traditional docking methods, can generate multiple plausible assembly structures, and achieves comparable performance to existing binary docking baselines, despite solving the harder task of coordinating multiple protein chains.
Adaptive Conformal Regression with Jackknife+ Rescaled Scores
May 31, 2023



Conformal regression provides prediction intervals with global coverage guarantees, but often fails to capture local error distributions, leading to non-homogeneous coverage. We address this with a new adaptive method based on rescaling conformal scores with an estimate of local score distribution, inspired by the Jackknife+ method, which enables the use of calibration data in conformal scores without breaking calibration-test exchangeability. Our approach ensures formal global coverage guarantees and is supported by new theoretical results on local coverage, including an a posteriori bound on any calibration score. The strength of our approach lies in achieving local coverage without sacrificing calibration set size, improving the applicability of conformal prediction intervals in various settings. As a result, our method provides prediction intervals that outperform previous methods, particularly in the low-data regime, making it especially relevant for real-world applications such as healthcare and biomedical domains where uncertainty needs to be quantified accurately despite low sample data.
Aligned Diffusion Schrödinger Bridges
Feb 22, 2023Diffusion Schr\"odinger bridges (DSB) have recently emerged as a powerful framework for recovering stochastic dynamics via their marginal observations at different time points. Despite numerous successful applications, existing algorithms for solving DSBs have so far failed to utilize the structure of aligned data, which naturally arises in many biological phenomena. In this paper, we propose a novel algorithmic framework that, for the first time, solves DSBs while respecting the data alignment. Our approach hinges on a combination of two decades-old ideas: The classical Schr\"odinger bridge theory and Doob's $h$-transform. Compared to prior methods, our approach leads to a simpler training procedure with lower variance, which we further augment with principled regularization schemes. This ultimately leads to sizeable improvements across experiments on synthetic and real data, including the tasks of rigid protein docking and temporal evolution of cellular differentiation processes.
It's FLAN time! Summing feature-wise latent representations for interpretability
Jun 18, 2021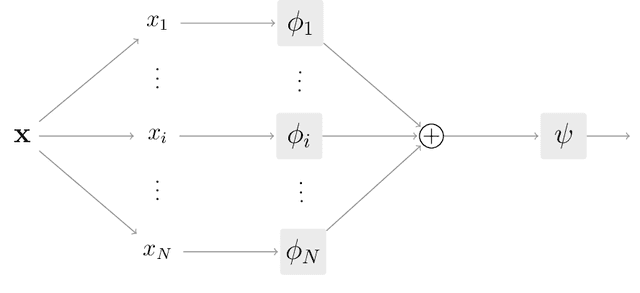
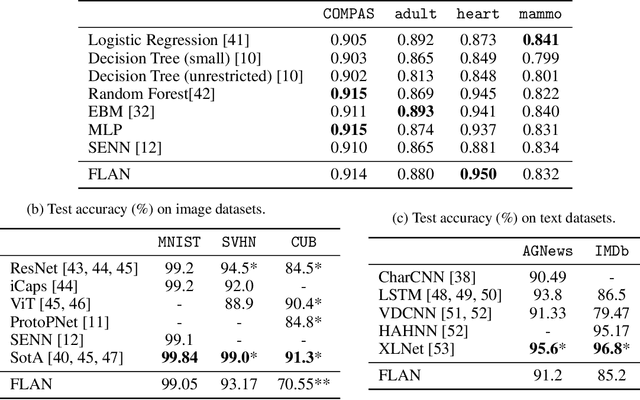
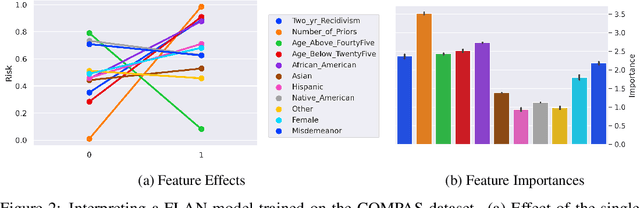
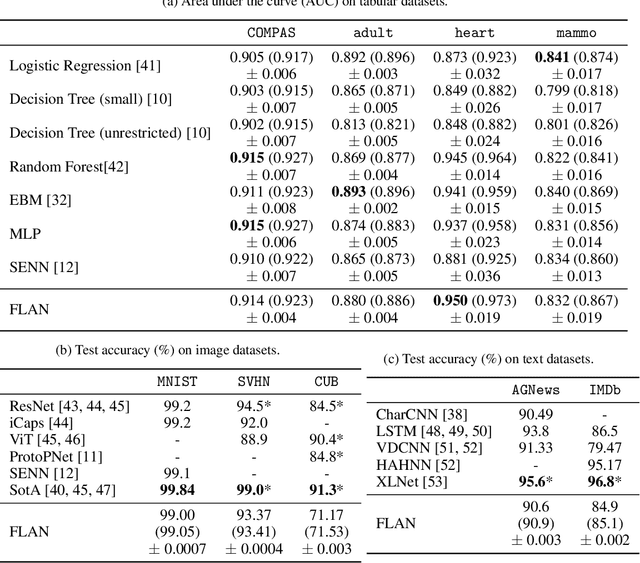
Interpretability has become a necessary feature for machine learning models deployed in critical scenarios, e.g. legal systems, healthcare. In these situations, algorithmic decisions may have (potentially negative) long-lasting effects on the end-user affected by the decision. In many cases, the representational power of deep learning models is not needed, therefore simple and interpretable models (e.g. linear models) should be preferred. However, in high-dimensional and/or complex domains (e.g. computer vision), the universal approximation capabilities of neural networks is required. Inspired by linear models and the Kolmogorov-Arnol representation theorem, we propose a novel class of structurally-constrained neural networks, which we call FLANs (Feature-wise Latent Additive Networks). Crucially, FLANs process each input feature separately, computing for each of them a representation in a common latent space. These feature-wise latent representations are then simply summed, and the aggregated representation is used for prediction. These constraints (which are at the core of the interpretability of linear models) allow an user to estimate the effect of each individual feature independently from the others, enhancing interpretability. In a set of experiments across different domains, we show how without compromising excessively the test performance, the structural constraints proposed in FLANs indeed increase the interpretability of deep learning models.
Pre-training Protein Language Models with Label-Agnostic Binding Pairs Enhances Performance in Downstream Tasks
Dec 05, 2020



Less than 1% of protein sequences are structurally and functionally annotated. Natural Language Processing (NLP) community has recently embraced self-supervised learning as a powerful approach to learn representations from unlabeled text, in large part due to the attention-based context-aware Transformer models. In this work we present a modification to the RoBERTa model by inputting during pre-training a mixture of binding and non-binding protein sequences (from STRING database). However, the sequence pairs have no label to indicate their binding status, as the model relies solely on Masked Language Modeling (MLM) objective during pre-training. After fine-tuning, such approach surpasses models trained on single protein sequences for protein-protein binding prediction, TCR-epitope binding prediction, cellular-localization and remote homology classification tasks. We suggest that the Transformer's attention mechanism contributes to protein binding site discovery. Furthermore, we compress protein sequences by 64% with the Byte Pair Encoding (BPE) vocabulary consisting of 10K subwords, each around 3-4 amino acids long. Finally, to expand the model input space to even larger proteins and multi-protein assemblies, we pre-train Longformer models that support 2,048 tokens. Further work in token-level classification for secondary structure prediction is needed. Code available at: https://github.com/PaccMann/paccmann_proteomics
Network-based Biased Tree Ensembles for Drug Sensitivity Prediction and Drug Sensitivity Biomarker Identification in Cancer
Aug 18, 2018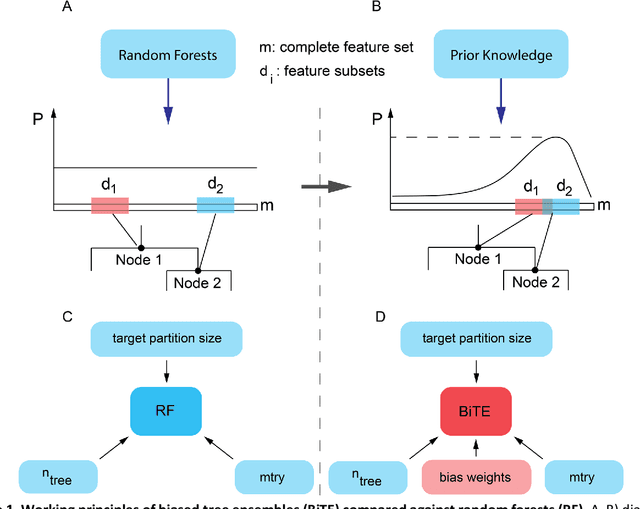
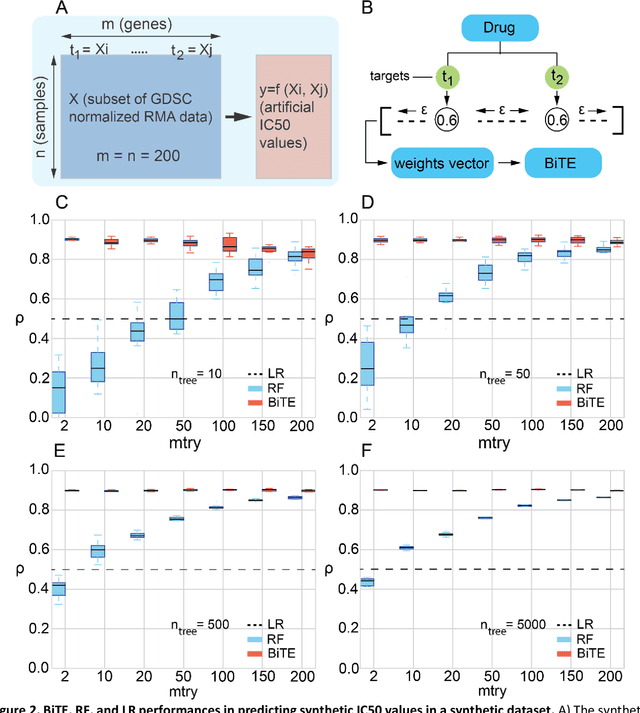
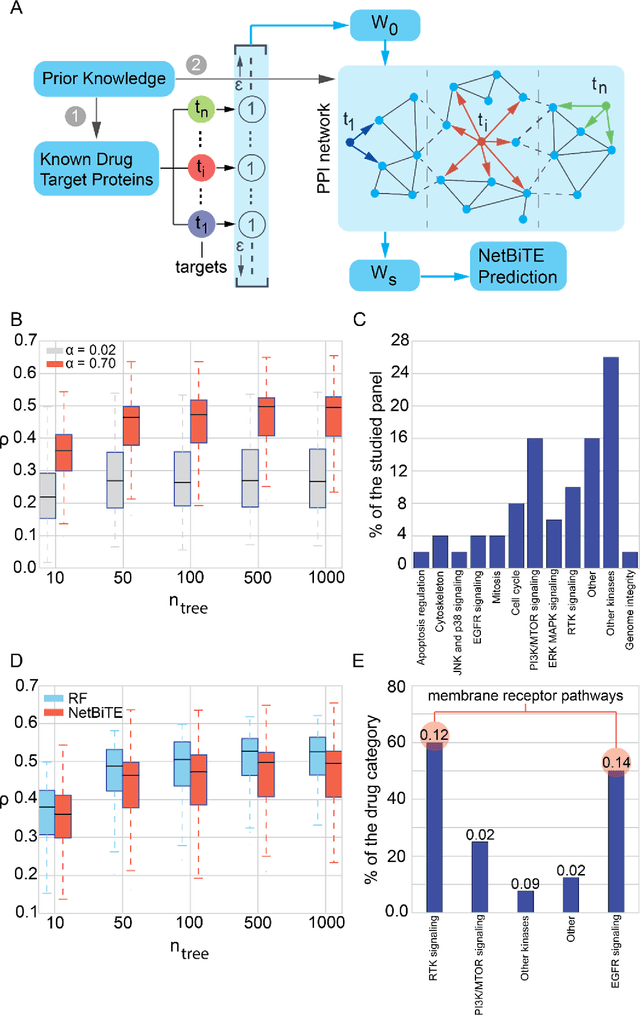
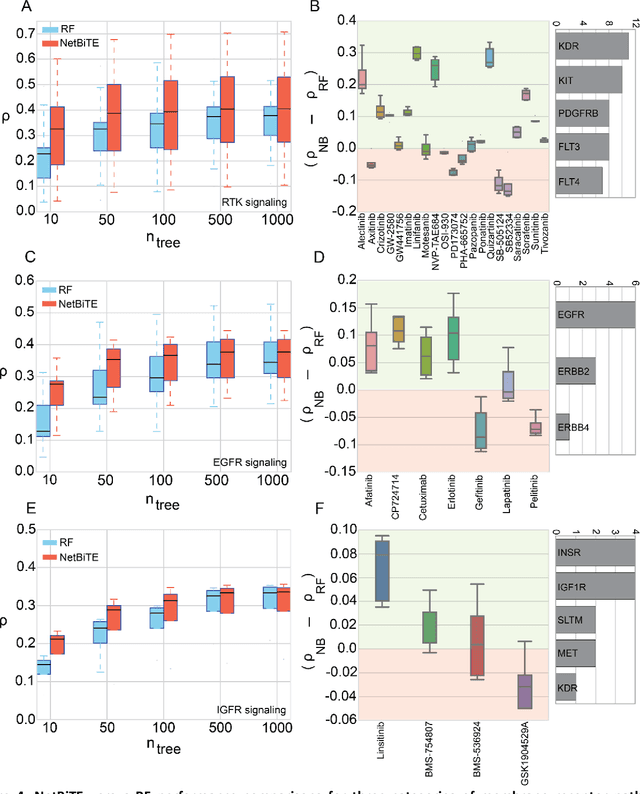
We present the Network-based Biased Tree Ensembles (NetBiTE) method for drug sensitivity prediction and drug sensitivity biomarker identification in cancer using a combination of prior knowledge and gene expression data. Our devised method consists of a biased tree ensemble that is built according to a probabilistic bias weight distribution. The bias weight distribution is obtained from the assignment of high weights to the drug targets and propagating the assigned weights over a protein-protein interaction network such as STRING. The propagation of weights, defines neighborhoods of influence around the drug targets and as such simulates the spread of perturbations within the cell, following drug administration. Using a synthetic dataset, we showcase how application of biased tree ensembles (BiTE) results in significant accuracy gains at a much lower computational cost compared to the unbiased random forests (RF) algorithm. We then apply NetBiTE to the Genomics of Drug Sensitivity in Cancer (GDSC) dataset and demonstrate that NetBiTE outperforms RF in predicting IC50 drug sensitivity, only for drugs that target membrane receptor pathways (MRPs): RTK, EGFR and IGFR signaling pathways. We propose based on the NetBiTE results, that for drugs that inhibit MRPs, the expression of target genes prior to drug administration is a biomarker for IC50 drug sensitivity following drug administration. We further verify and reinforce this proposition through control studies on, PI3K/MTOR signaling pathway inhibitors, a drug category that does not target MRPs, and through assignment of dummy targets to MRP inhibiting drugs and investigating the variation in NetBiTE accuracy.