 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeReliable LLM-based User Simulator for Task-Oriented Dialogue Systems
Feb 20, 2024



In the realm of dialogue systems, user simulation techniques have emerged as a game-changer, redefining the evaluation and enhancement of task-oriented dialogue (TOD) systems. These methods are crucial for replicating real user interactions, enabling applications like synthetic data augmentation, error detection, and robust evaluation. However, existing approaches often rely on rigid rule-based methods or on annotated data. This paper introduces DAUS, a Domain-Aware User Simulator. Leveraging large language models, we fine-tune DAUS on real examples of task-oriented dialogues. Results on two relevant benchmarks showcase significant improvements in terms of user goal fulfillment. Notably, we have observed that fine-tuning enhances the simulator's coherence with user goals, effectively mitigating hallucinations -- a major source of inconsistencies in simulator responses.
In-Context Learning User Simulators for Task-Oriented Dialog Systems
Jun 01, 2023



This paper presents a novel application of large language models in user simulation for task-oriented dialog systems, specifically focusing on an in-context learning approach. By harnessing the power of these models, the proposed approach generates diverse utterances based on user goals and limited dialog examples. Unlike traditional simulators, this method eliminates the need for labor-intensive rule definition or extensive annotated data, making it more efficient and accessible. Additionally, an error analysis of the interaction between the user simulator and dialog system uncovers common mistakes, providing valuable insights into areas that require improvement. Our implementation is available at https://github.com/telepathylabsai/prompt-based-user-simulator.
Pre-training Protein Language Models with Label-Agnostic Binding Pairs Enhances Performance in Downstream Tasks
Dec 05, 2020



Less than 1% of protein sequences are structurally and functionally annotated. Natural Language Processing (NLP) community has recently embraced self-supervised learning as a powerful approach to learn representations from unlabeled text, in large part due to the attention-based context-aware Transformer models. In this work we present a modification to the RoBERTa model by inputting during pre-training a mixture of binding and non-binding protein sequences (from STRING database). However, the sequence pairs have no label to indicate their binding status, as the model relies solely on Masked Language Modeling (MLM) objective during pre-training. After fine-tuning, such approach surpasses models trained on single protein sequences for protein-protein binding prediction, TCR-epitope binding prediction, cellular-localization and remote homology classification tasks. We suggest that the Transformer's attention mechanism contributes to protein binding site discovery. Furthermore, we compress protein sequences by 64% with the Byte Pair Encoding (BPE) vocabulary consisting of 10K subwords, each around 3-4 amino acids long. Finally, to expand the model input space to even larger proteins and multi-protein assemblies, we pre-train Longformer models that support 2,048 tokens. Further work in token-level classification for secondary structure prediction is needed. Code available at: https://github.com/PaccMann/paccmann_proteomics
PaccMann$^{RL}$ on SARS-CoV-2: Designing antiviral candidates with conditional generative models
May 31, 2020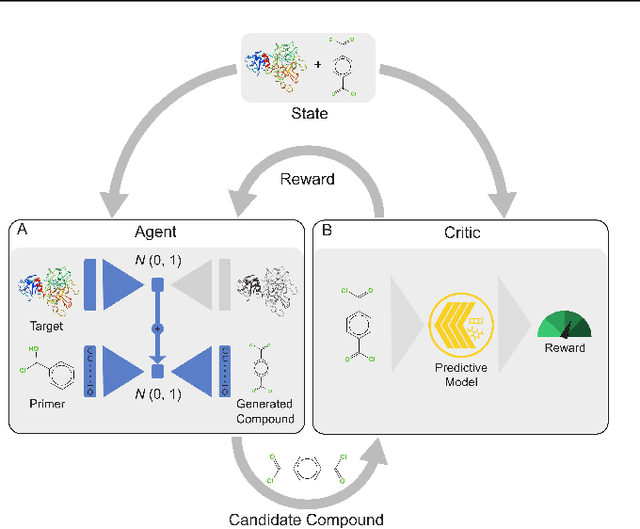

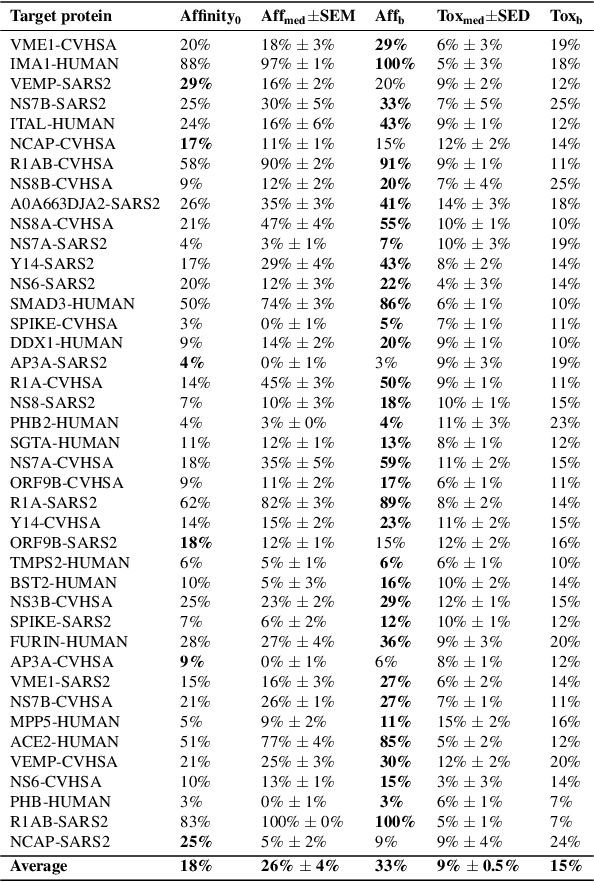
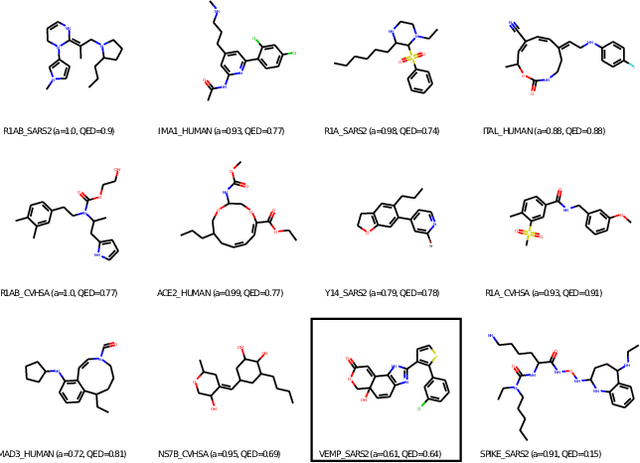
With the fast development of COVID-19 into a global pandemic, scientists around the globe are desperately searching for effective antiviral therapeutic agents. Bridging systems biology and drug discovery, we propose a deep learning framework for conditional de novo design of antiviral candidate drugs tailored against given protein targets. First, we train a multimodal ligand--protein binding affinity model on predicting affinities of antiviral compounds to target proteins and couple this model with pharmacological toxicity predictors. Exploiting this multi-objective as a reward function of a conditional molecular generator (consisting of two VAEs), we showcase a framework that navigates the chemical space toward regions with more antiviral molecules. Specifically, we explore a challenging setting of generating ligands against unseen protein targets by performing a leave-one-out-cross-validation on 41 SARS-CoV-2-related target proteins. Using deep RL, it is demonstrated that in 35 out of 41 cases, the generation is biased towards sampling more binding ligands, with an average increase of 83% comparing to an unbiased VAE. We present a case-study on a potential Envelope-protein inhibitor and perform a synthetic accessibility assessment of the best generated molecules is performed that resembles a viable roadmap towards a rapid in-vitro evaluation of potential SARS-CoV-2 inhibitors.