 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUnlearning Information Bottleneck: Machine Unlearning of Systematic Patterns and Biases
May 22, 2024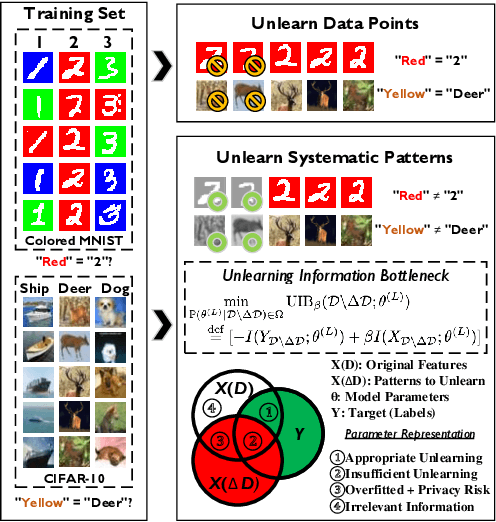
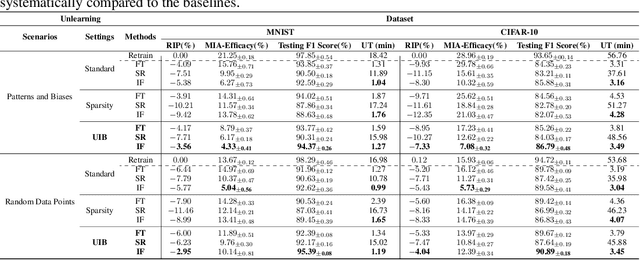
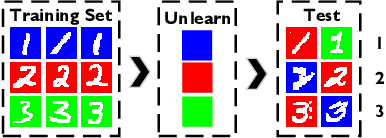

Effective adaptation to distribution shifts in training data is pivotal for sustaining robustness in neural networks, especially when removing specific biases or outdated information, a process known as machine unlearning. Traditional approaches typically assume that data variations are random, which makes it difficult to adjust the model parameters accurately to remove patterns and characteristics from unlearned data. In this work, we present Unlearning Information Bottleneck (UIB), a novel information-theoretic framework designed to enhance the process of machine unlearning that effectively leverages the influence of systematic patterns and biases for parameter adjustment. By proposing a variational upper bound, we recalibrate the model parameters through a dynamic prior that integrates changes in data distribution with an affordable computational cost, allowing efficient and accurate removal of outdated or unwanted data patterns and biases. Our experiments across various datasets, models, and unlearning methods demonstrate that our approach effectively removes systematic patterns and biases while maintaining the performance of models post-unlearning.
Conformal Autoregressive Generation: Beam Search with Coverage Guarantees
Sep 07, 2023We introduce two new extensions to the beam search algorithm based on conformal predictions (CP) to produce sets of sequences with theoretical coverage guarantees. The first method is very simple and proposes dynamically-sized subsets of beam search results but, unlike typical CP procedures, has an upper bound on the achievable guarantee depending on a post-hoc calibration measure. Our second algorithm introduces the conformal set prediction procedure as part of the decoding process, producing a variable beam width which adapts to the current uncertainty. While more complex, this procedure can achieve coverage guarantees selected a priori. We provide marginal coverage bounds for each method, and evaluate them empirically on a selection of tasks drawing from natural language processing and chemistry.
Attention-based Interpretable Regression of Gene Expression in Histology
Aug 29, 2022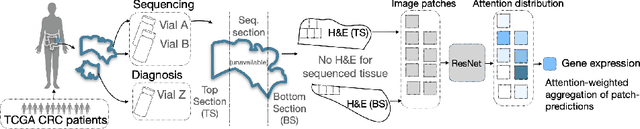
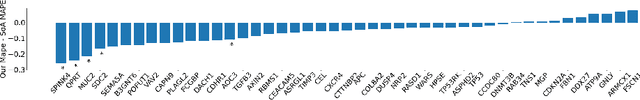


Interpretability of deep learning is widely used to evaluate the reliability of medical imaging models and reduce the risks of inaccurate patient recommendations. For models exceeding human performance, e.g. predicting RNA structure from microscopy images, interpretable modelling can be further used to uncover highly non-trivial patterns which are otherwise imperceptible to the human eye. We show that interpretability can reveal connections between the microscopic appearance of cancer tissue and its gene expression profiling. While exhaustive profiling of all genes from the histology images is still challenging, we estimate the expression values of a well-known subset of genes that is indicative of cancer molecular subtype, survival, and treatment response in colorectal cancer. Our approach successfully identifies meaningful information from the image slides, highlighting hotspots of high gene expression. Our method can help characterise how gene expression shapes tissue morphology and this may be beneficial for patient stratification in the pathology unit. The code is available on GitHub.
TITAN: T Cell Receptor Specificity Prediction with Bimodal Attention Networks
Apr 21, 2021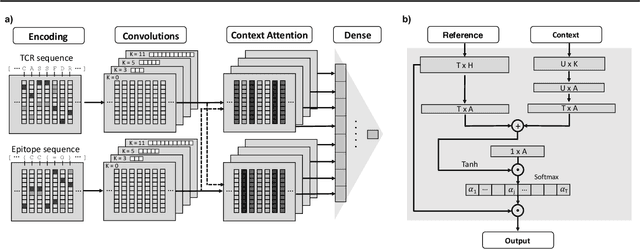

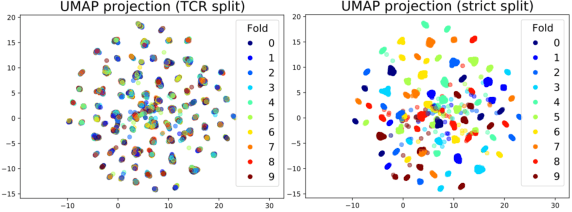
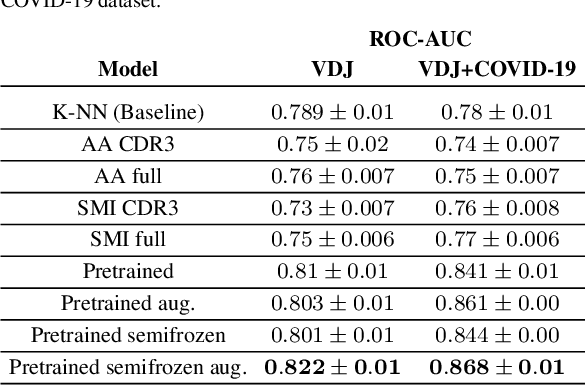
Motivation: The activity of the adaptive immune system is governed by T-cells and their specific T-cell receptors (TCR), which selectively recognize foreign antigens. Recent advances in experimental techniques have enabled sequencing of TCRs and their antigenic targets (epitopes), allowing to research the missing link between TCR sequence and epitope binding specificity. Scarcity of data and a large sequence space make this task challenging, and to date only models limited to a small set of epitopes have achieved good performance. Here, we establish a k-nearest-neighbor (K-NN) classifier as a strong baseline and then propose TITAN (Tcr epITope bimodal Attention Networks), a bimodal neural network that explicitly encodes both TCR sequences and epitopes to enable the independent study of generalization capabilities to unseen TCRs and/or epitopes. Results: By encoding epitopes at the atomic level with SMILES sequences, we leverage transfer learning and data augmentation to enrich the input data space and boost performance. TITAN achieves high performance in the prediction of specificity of unseen TCRs (ROC-AUC 0.87 in 10-fold CV) and surpasses the results of the current state-of-the-art (ImRex) by a large margin. Notably, our Levenshtein-distance-based K-NN classifier also exhibits competitive performance on unseen TCRs. While the generalization to unseen epitopes remains challenging, we report two major breakthroughs. First, by dissecting the attention heatmaps, we demonstrate that the sparsity of available epitope data favors an implicit treatment of epitopes as classes. This may be a general problem that limits unseen epitope performance for sufficiently complex models. Second, we show that TITAN nevertheless exhibits significantly improved performance on unseen epitopes and is capable of focusing attention on chemically meaningful molecular structures.
Learning Invariances for Interpretability using Supervised VAE
Jul 15, 2020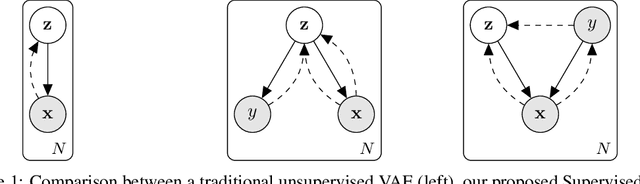

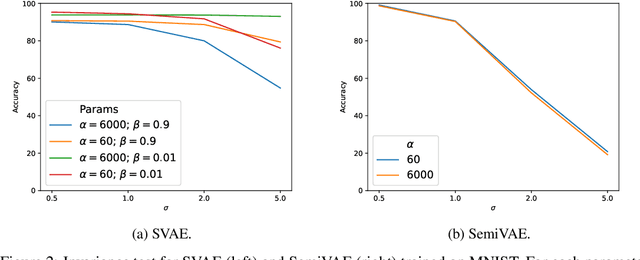

We propose to learn model invariances as a means of interpreting a model. This is motivated by a reverse engineering principle. If we understand a problem, we may introduce inductive biases in our model in the form of invariances. Conversely, when interpreting a complex supervised model, we can study its invariances to understand how that model solves a problem. To this end we propose a supervised form of variational auto-encoders (VAEs). Crucially, only a subset of the dimensions in the latent space contributes to the supervised task, allowing the remaining dimensions to act as nuisance parameters. By sampling solely the nuisance dimensions, we are able to generate samples that have undergone transformations that leave the classification unchanged, revealing the invariances of the model. Our experimental results show the capability of our proposed model both in terms of classification, and generation of invariantly transformed samples. Finally we show how combining our model with feature attribution methods it is possible to reach a more fine-grained understanding about the decision process of the model.
On quantitative aspects of model interpretability
Jul 15, 2020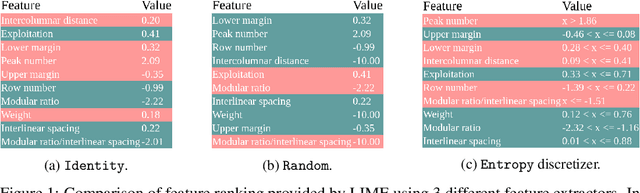
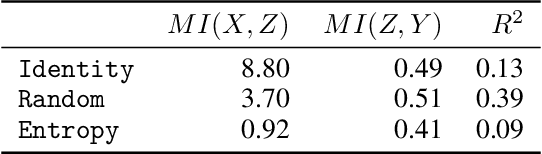

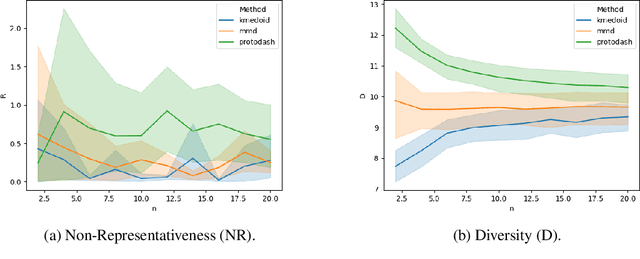
Despite the growing body of work in interpretable machine learning, it remains unclear how to evaluate different explainability methods without resorting to qualitative assessment and user-studies. While interpretability is an inherently subjective matter, previous works in cognitive science and epistemology have shown that good explanations do possess aspects that can be objectively judged apart from fidelity), such assimplicity and broadness. In this paper we propose a set of metrics to programmatically evaluate interpretability methods along these dimensions. In particular, we argue that the performance of methods along these dimensions can be orthogonally imputed to two conceptual parts, namely the feature extractor and the actual explainability method. We experimentally validate our metrics on different benchmark tasks and show how they can be used to guide a practitioner in the selection of the most appropriate method for the task at hand.
PaccMann$^{RL}$ on SARS-CoV-2: Designing antiviral candidates with conditional generative models
May 31, 2020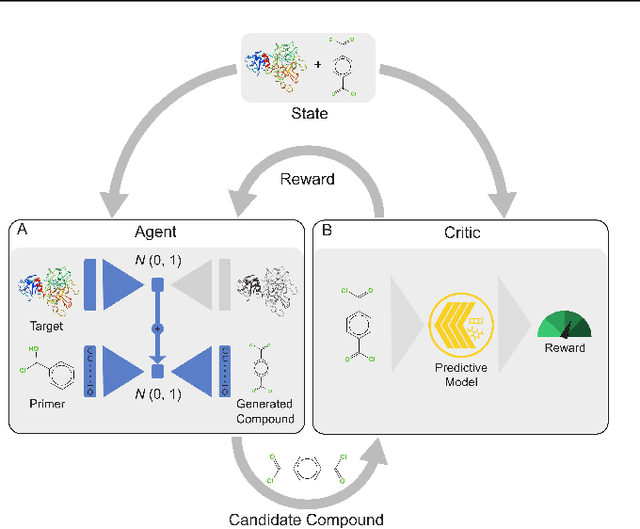

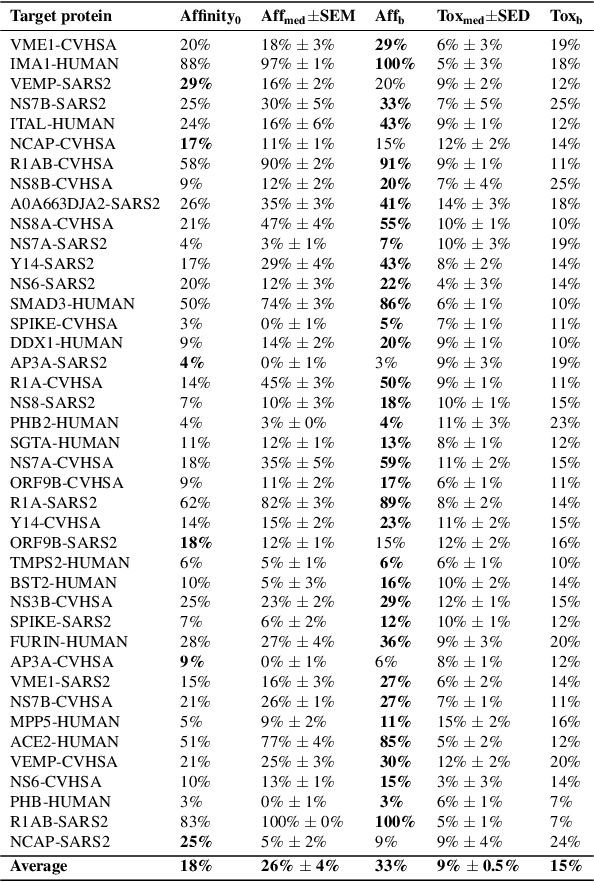
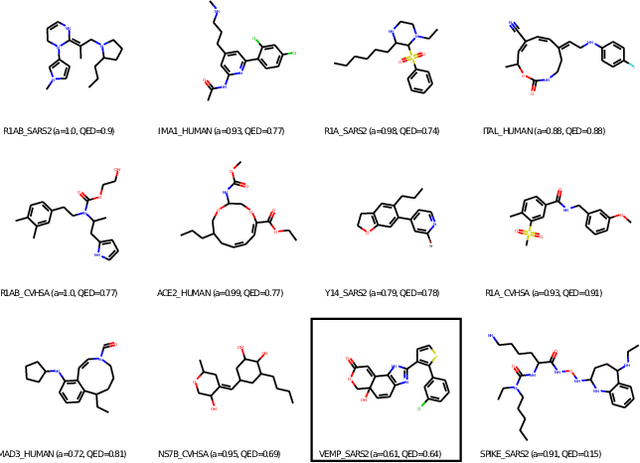
With the fast development of COVID-19 into a global pandemic, scientists around the globe are desperately searching for effective antiviral therapeutic agents. Bridging systems biology and drug discovery, we propose a deep learning framework for conditional de novo design of antiviral candidate drugs tailored against given protein targets. First, we train a multimodal ligand--protein binding affinity model on predicting affinities of antiviral compounds to target proteins and couple this model with pharmacological toxicity predictors. Exploiting this multi-objective as a reward function of a conditional molecular generator (consisting of two VAEs), we showcase a framework that navigates the chemical space toward regions with more antiviral molecules. Specifically, we explore a challenging setting of generating ligands against unseen protein targets by performing a leave-one-out-cross-validation on 41 SARS-CoV-2-related target proteins. Using deep RL, it is demonstrated that in 35 out of 41 cases, the generation is biased towards sampling more binding ligands, with an average increase of 83% comparing to an unbiased VAE. We present a case-study on a potential Envelope-protein inhibitor and perform a synthetic accessibility assessment of the best generated molecules is performed that resembles a viable roadmap towards a rapid in-vitro evaluation of potential SARS-CoV-2 inhibitors.
DeStress: Deep Learning for Unsupervised Identification of Mental Stress in Firefighters from Heart-rate Variability (HRV) Data
Nov 18, 2019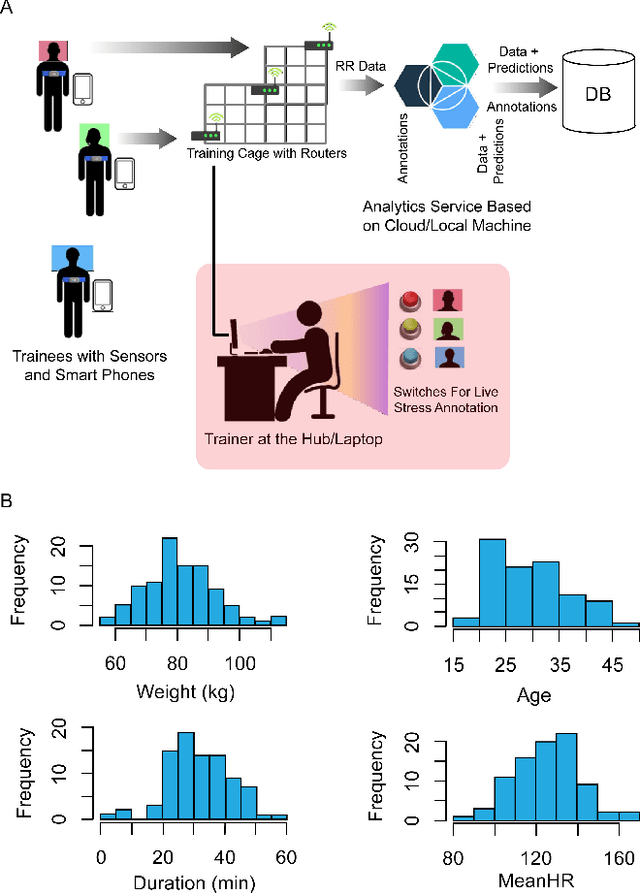
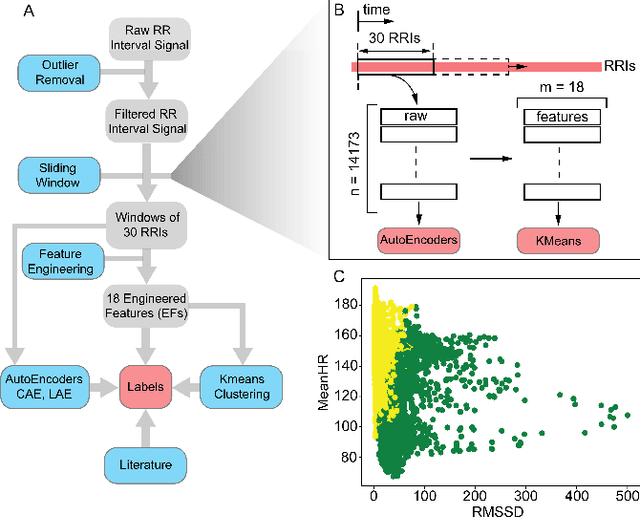


In this work we perform a study of various unsupervised methods to identify mental stress in firefighter trainees based on unlabeled heart rate variability data. We collect RR interval time series data from nearly 100 firefighter trainees that participated in a drill. We explore and compare three methods in order to perform unsupervised stress detection: 1) traditional K-Means clustering with engineered time and frequency domain features 2) convolutional autoencoders and 3) long short-term memory (LSTM) autoencoders, both trained on the raw RRI measurements combined with DBSCAN clustering and K-Nearest-Neighbors classification. We demonstrate that K-Means combined with engineered features is unable to capture meaningful structure within the data. On the other hand, convolutional and LSTM autoencoders tend to extract varying structure from the data pointing to different clusters with different sizes of clusters. We attempt at identifying the true stressed and normal clusters using the HRV markers of mental stress reported in the literature. We demonstrate that the clusters produced by the convolutional autoencoders consistently and successfully stratify stressed versus normal samples, as validated by several established physiological stress markers such as RMSSD, Max-HR, Mean-HR and LF-HF ratio.
MonoNet: Towards Interpretable Models by Learning Monotonic Features
Sep 30, 2019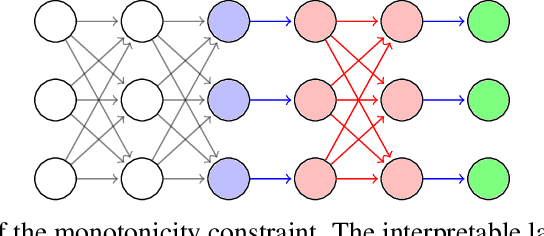



Being able to interpret, or explain, the predictions made by a machine learning model is of fundamental importance. This is especially true when there is interest in deploying data-driven models to make high-stakes decisions, e.g. in healthcare. While recent years have seen an increasing interest in interpretable machine learning research, this field is currently lacking an agreed-upon definition of interpretability, and some researchers have called for a more active conversation towards a rigorous approach to interpretability. Joining this conversation, we claim in this paper that the difficulty of interpreting a complex model stems from the existing interactions among features. We argue that by enforcing monotonicity between features and outputs, we are able to reason about the effect of a single feature on an output independently from other features, and consequently better understand the model. We show how to structurally introduce this constraint in deep learning models by adding new simple layers. We validate our model on benchmark datasets, and compare our results with previously proposed interpretable models.
Reinforcement learning-driven de-novo design of anticancer compounds conditioned on biomolecular profiles
Aug 29, 2019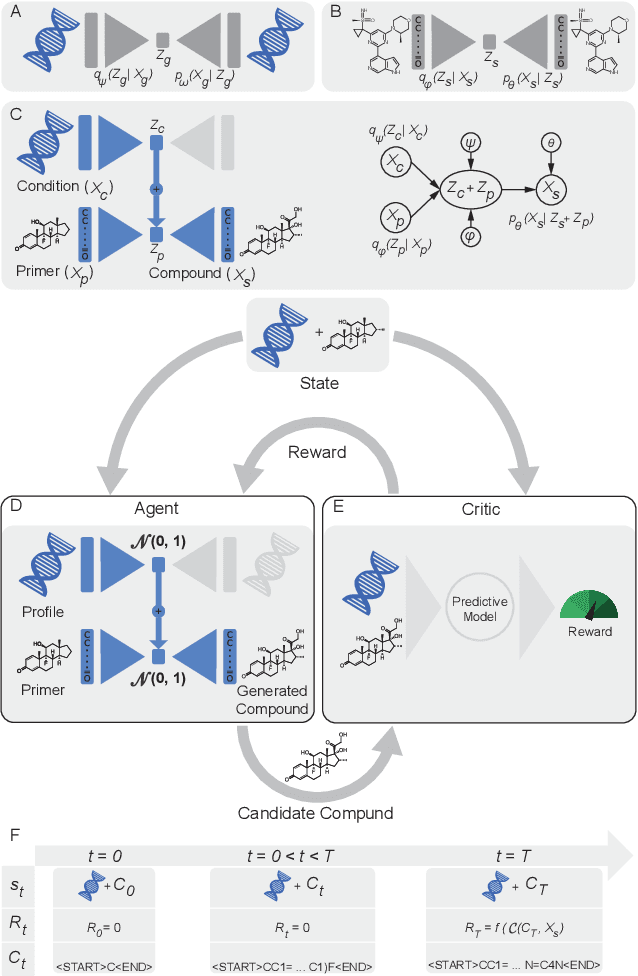
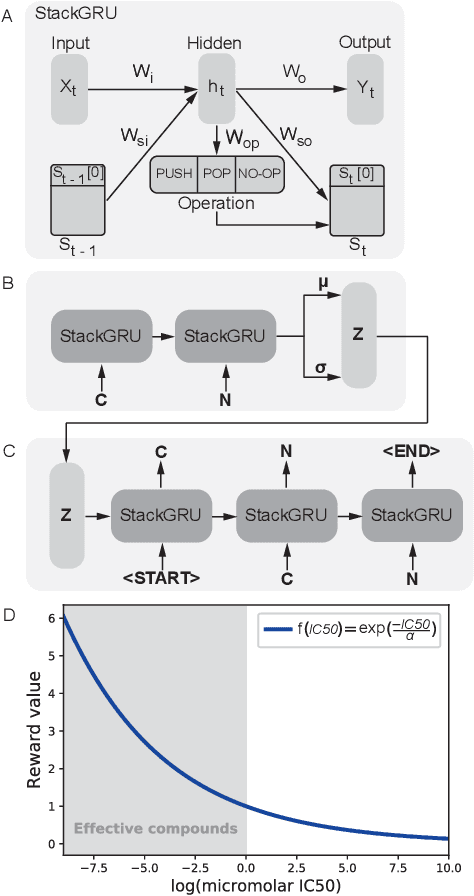
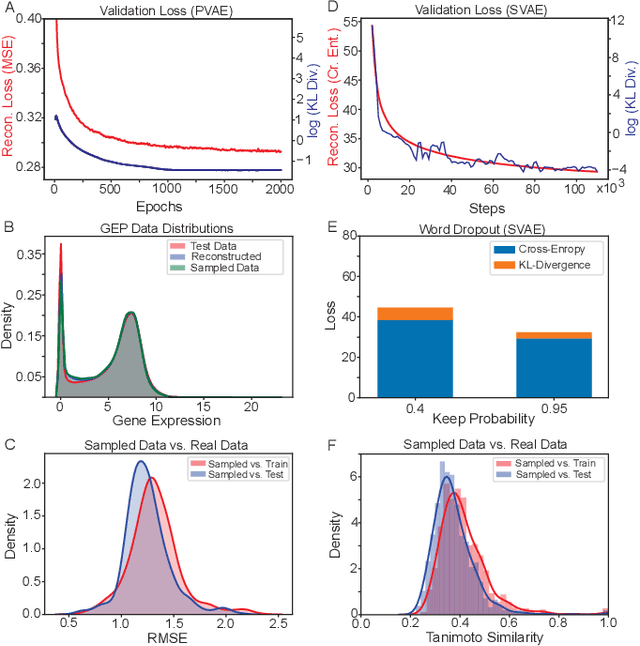
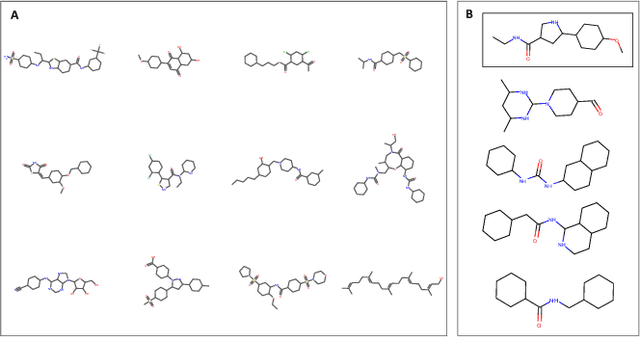
With the advent of deep generative models in computational chemistry, in silico anticancer drug design has undergone an unprecedented transformation. While state-of-the-art deep learning approaches have shown potential in generating compounds with desired chemical properties, they entirely overlook the genetic profile and properties of the target disease. In the case of cancer, this is problematic since it is a highly genetic disease in which the biomolecular profile of target cells determines the response to therapy. Here, we introduce the first deep generative model capable of generating anticancer compounds given a target biomolecular profile. Using a reinforcement learning framework, the transcriptomic profile of cancer cells is used as a context in which anticancer molecules are generated and optimized to obtain effective compounds for the given profile. Our molecule generator combines two pretrained variational autoencoders (VAEs) and a multimodal efficacy predictor - the first VAE generates transcriptomic profiles while the second conditional VAE generates novel molecular structures conditioned on the given transcriptomic profile. The efficacy predictor is used to optimize the generated molecules through a reward determined by the predicted IC50 drug sensitivity for the generated molecule and the target profile. We demonstrate how the molecule generation can be biased towards compounds with high inhibitory effect against individual cell lines or specific cancer sites. We verify our approach by investigating candidate drugs generated against specific cancer types and investigate their structural similarity to existing compounds with known efficacy against these cancer types. We envision our approach to transform in silico anticancer drug design by increasing success rates in lead compound discovery via leveraging the biomolecular characteristics of the disease.