 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNamed Entity Recognition in the Legal Domain using a Pointer Generator Network
Dec 17, 2020

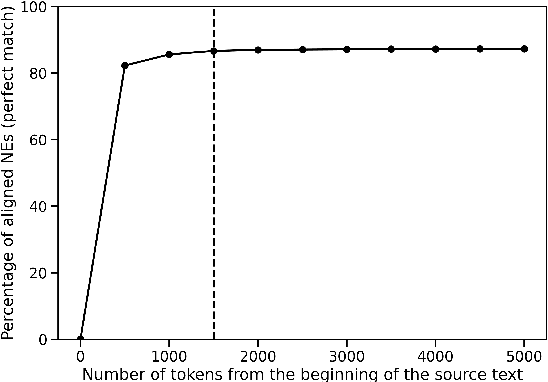
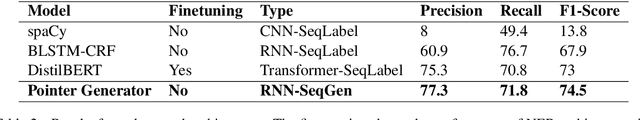
Named Entity Recognition (NER) is the task of identifying and classifying named entities in unstructured text. In the legal domain, named entities of interest may include the case parties, judges, names of courts, case numbers, references to laws etc. We study the problem of legal NER with noisy text extracted from PDF files of filed court cases from US courts. The "gold standard" training data for NER systems provide annotation for each token of the text with the corresponding entity or non-entity label. We work with only partially complete training data, which differ from the gold standard NER data in that the exact location of the entities in the text is unknown and the entities may contain typos and/or OCR mistakes. To overcome the challenges of our noisy training data, e.g. text extraction errors and/or typos and unknown label indices, we formulate the NER task as a text-to-text sequence generation task and train a pointer generator network to generate the entities in the document rather than label them. We show that the pointer generator can be effective for NER in the absence of gold standard data and outperforms the common NER neural network architectures in long legal documents.
DeStress: Deep Learning for Unsupervised Identification of Mental Stress in Firefighters from Heart-rate Variability (HRV) Data
Nov 18, 2019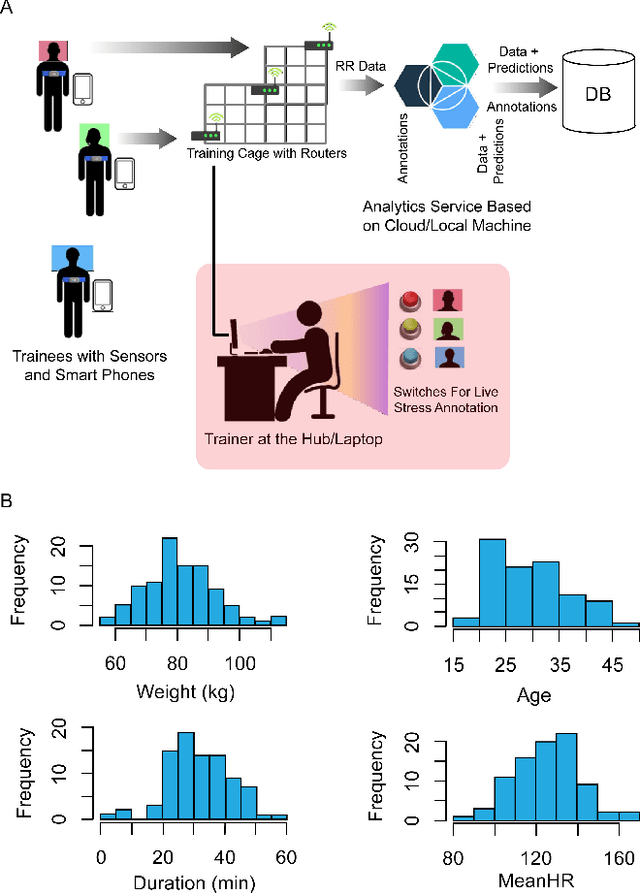
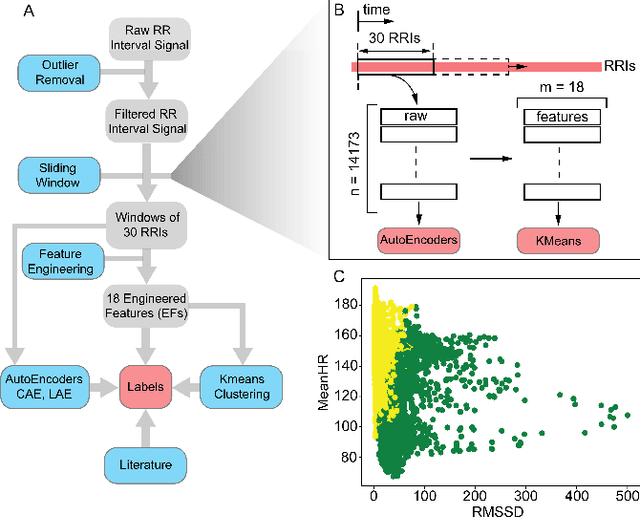


In this work we perform a study of various unsupervised methods to identify mental stress in firefighter trainees based on unlabeled heart rate variability data. We collect RR interval time series data from nearly 100 firefighter trainees that participated in a drill. We explore and compare three methods in order to perform unsupervised stress detection: 1) traditional K-Means clustering with engineered time and frequency domain features 2) convolutional autoencoders and 3) long short-term memory (LSTM) autoencoders, both trained on the raw RRI measurements combined with DBSCAN clustering and K-Nearest-Neighbors classification. We demonstrate that K-Means combined with engineered features is unable to capture meaningful structure within the data. On the other hand, convolutional and LSTM autoencoders tend to extract varying structure from the data pointing to different clusters with different sizes of clusters. We attempt at identifying the true stressed and normal clusters using the HRV markers of mental stress reported in the literature. We demonstrate that the clusters produced by the convolutional autoencoders consistently and successfully stratify stressed versus normal samples, as validated by several established physiological stress markers such as RMSSD, Max-HR, Mean-HR and LF-HF ratio.
Reinforcement learning-driven de-novo design of anticancer compounds conditioned on biomolecular profiles
Aug 29, 2019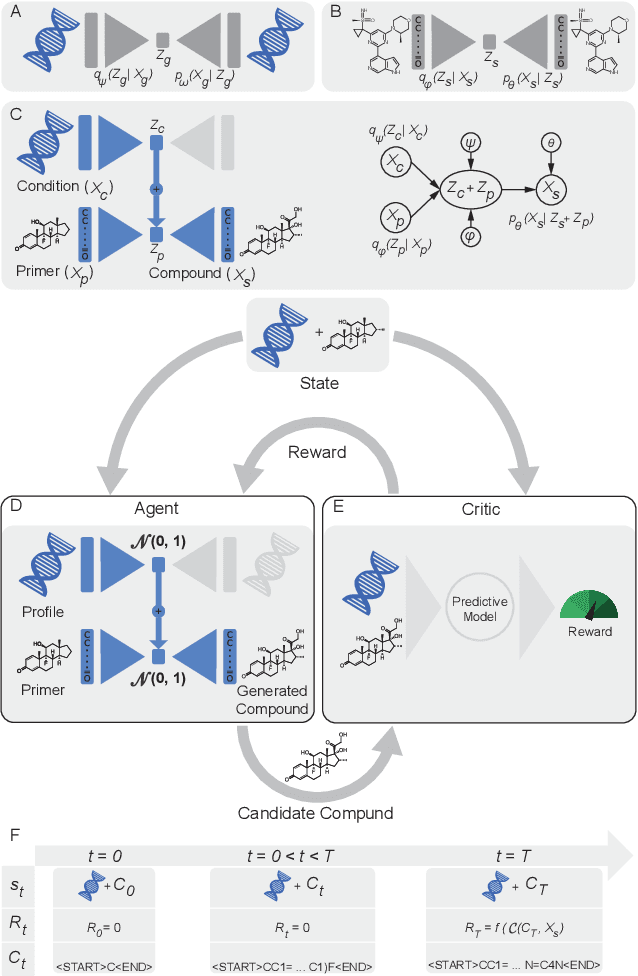
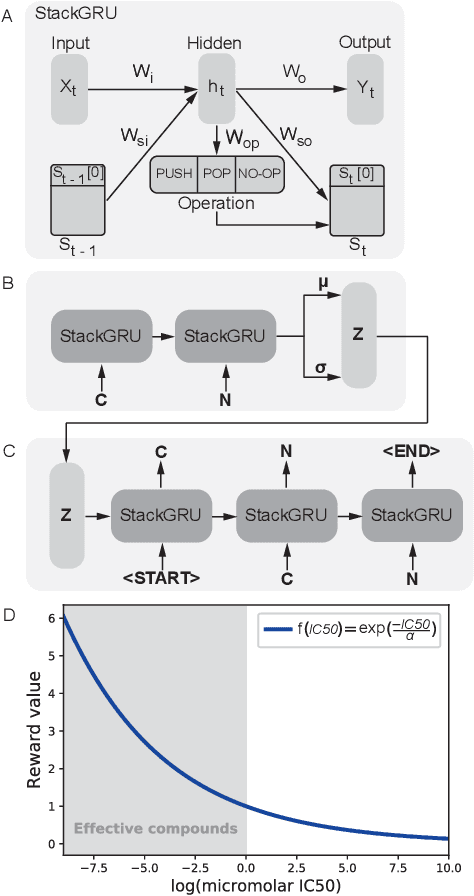
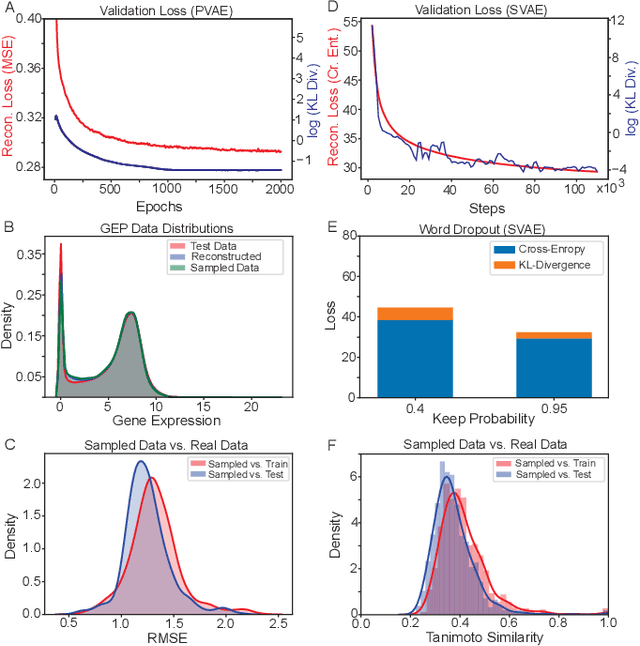
With the advent of deep generative models in computational chemistry, in silico anticancer drug design has undergone an unprecedented transformation. While state-of-the-art deep learning approaches have shown potential in generating compounds with desired chemical properties, they entirely overlook the genetic profile and properties of the target disease. In the case of cancer, this is problematic since it is a highly genetic disease in which the biomolecular profile of target cells determines the response to therapy. Here, we introduce the first deep generative model capable of generating anticancer compounds given a target biomolecular profile. Using a reinforcement learning framework, the transcriptomic profile of cancer cells is used as a context in which anticancer molecules are generated and optimized to obtain effective compounds for the given profile. Our molecule generator combines two pretrained variational autoencoders (VAEs) and a multimodal efficacy predictor - the first VAE generates transcriptomic profiles while the second conditional VAE generates novel molecular structures conditioned on the given transcriptomic profile. The efficacy predictor is used to optimize the generated molecules through a reward determined by the predicted IC50 drug sensitivity for the generated molecule and the target profile. We demonstrate how the molecule generation can be biased towards compounds with high inhibitory effect against individual cell lines or specific cancer sites. We verify our approach by investigating candidate drugs generated against specific cancer types and investigate their structural similarity to existing compounds with known efficacy against these cancer types. We envision our approach to transform in silico anticancer drug design by increasing success rates in lead compound discovery via leveraging the biomolecular characteristics of the disease.
Towards Explainable Anticancer Compound Sensitivity Prediction via Multimodal Attention-based Convolutional Encoders
May 22, 2019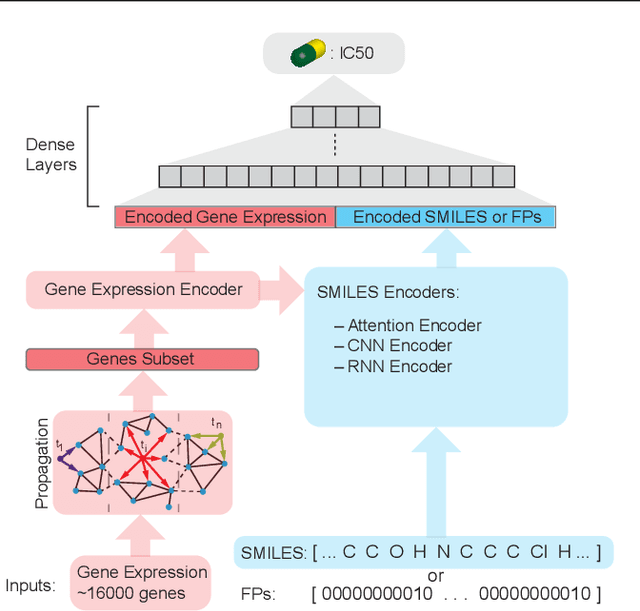
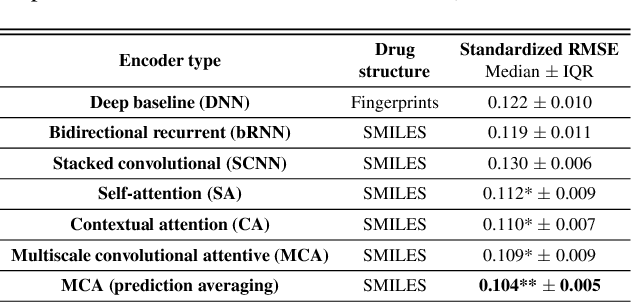
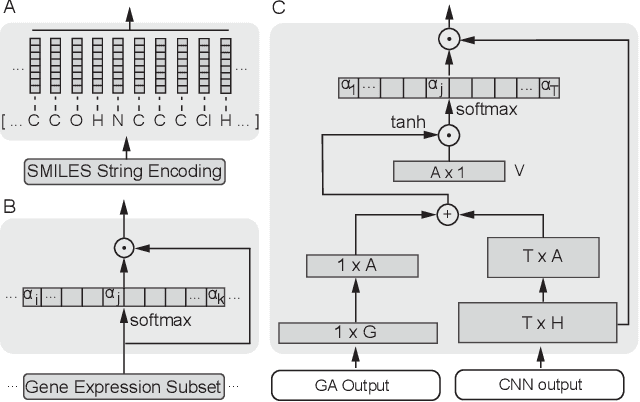
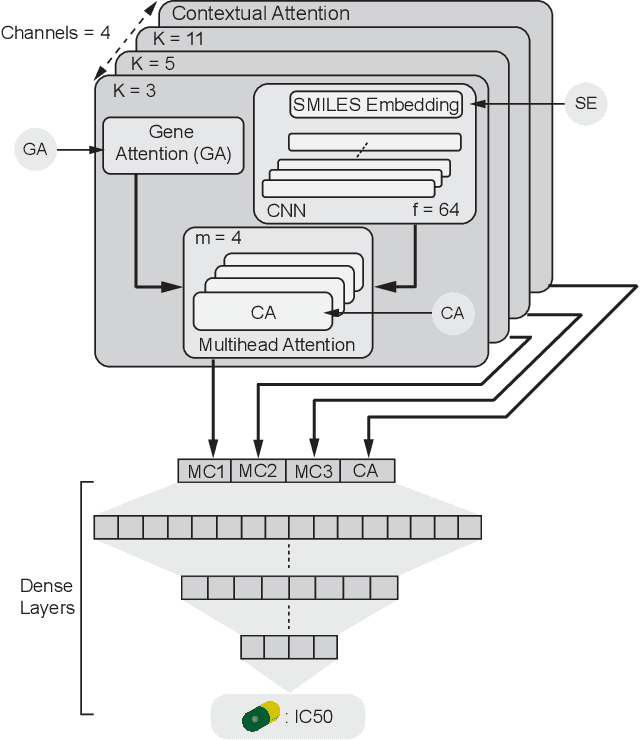
In line with recent advances in neural drug design and sensitivity prediction, we propose a novel architecture for interpretable prediction of anticancer compound sensitivity using a multimodal attention-based convolutional encoder. Our model is based on the three key pillars of drug sensitivity: compounds' structure in the form of a SMILES sequence, gene expression profiles of tumors and prior knowledge on intracellular interactions from protein-protein interaction networks. We demonstrate that our multiscale convolutional attention-based (MCA) encoder significantly outperforms a baseline model trained on Morgan fingerprints, a selection of encoders based on SMILES as well as previously reported state of the art for multimodal drug sensitivity prediction (R2 = 0.86 and RMSE = 0.89). Moreover, the explainability of our approach is demonstrated by a thorough analysis of the attention weights. We show that the attended genes significantly enrich apoptotic processes and that the drug attention is strongly correlated with a standard chemical structure similarity index. Finally, we report a case study of two receptor tyrosine kinase (RTK) inhibitors acting on a leukemia cell line, showcasing the ability of the model to focus on informative genes and submolecular regions of the two compounds. The demonstrated generalizability and the interpretability of our model testify its potential for in-silico prediction of anticancer compound efficacy on unseen cancer cells, positioning it as a valid solution for the development of personalized therapies as well as for the evaluation of candidate compounds in de novo drug design.
PaccMann: Prediction of anticancer compound sensitivity with multi-modal attention-based neural networks
Nov 16, 2018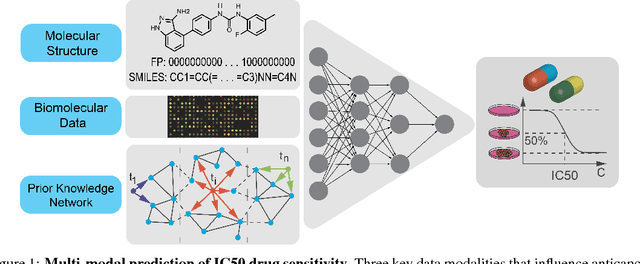
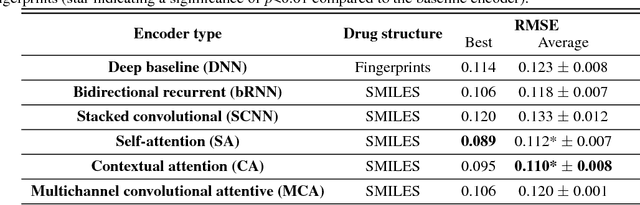
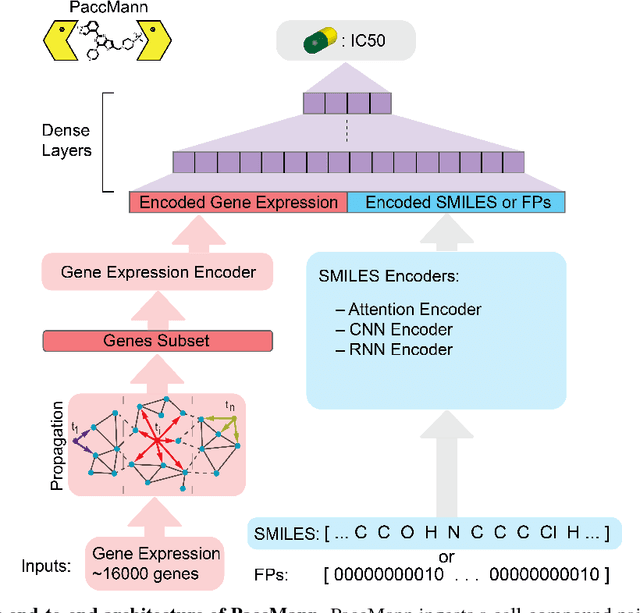
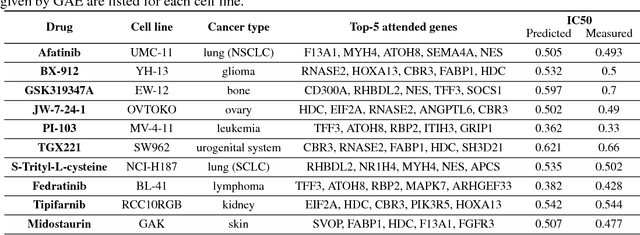
We present a novel approach for the prediction of anticancer compound sensitivity by means of multi-modal attention-based neural networks (PaccMann). In our approach, we integrate three key pillars of drug sensitivity, namely, the molecular structure of compounds, transcriptomic profiles of cancer cells as well as prior knowledge about interactions among proteins within cells. Our models ingest a drug-cell pair consisting of SMILES encoding of a compound and the gene expression profile of a cancer cell and predicts an IC50 sensitivity value. Gene expression profiles are encoded using an attention-based encoding mechanism that assigns high weights to the most informative genes. We present and study three encoders for SMILES string of compounds: 1) bidirectional recurrent 2) convolutional 3) attention-based encoders. We compare our devised models against a baseline model that ingests engineered fingerprints to represent the molecular structure. We demonstrate that using our attention-based encoders, we can surpass the baseline model. The use of attention-based encoders enhance interpretability and enable us to identify genes, bonds and atoms that were used by the network to make a prediction.
Network-based Biased Tree Ensembles for Drug Sensitivity Prediction and Drug Sensitivity Biomarker Identification in Cancer
Aug 18, 2018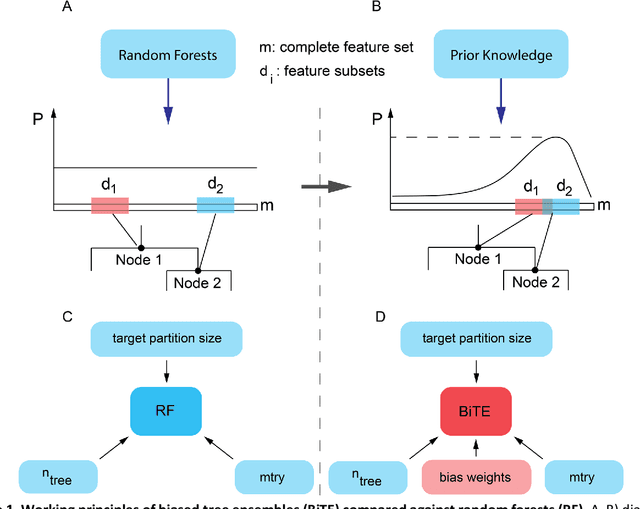
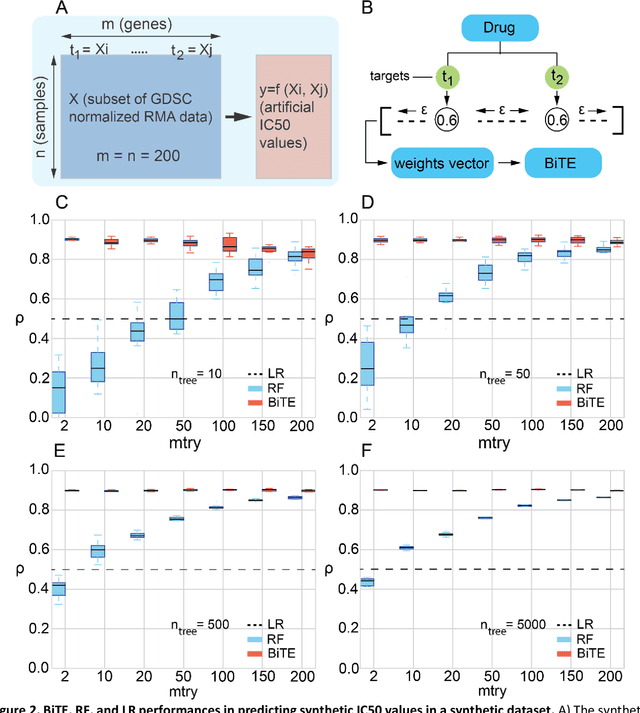
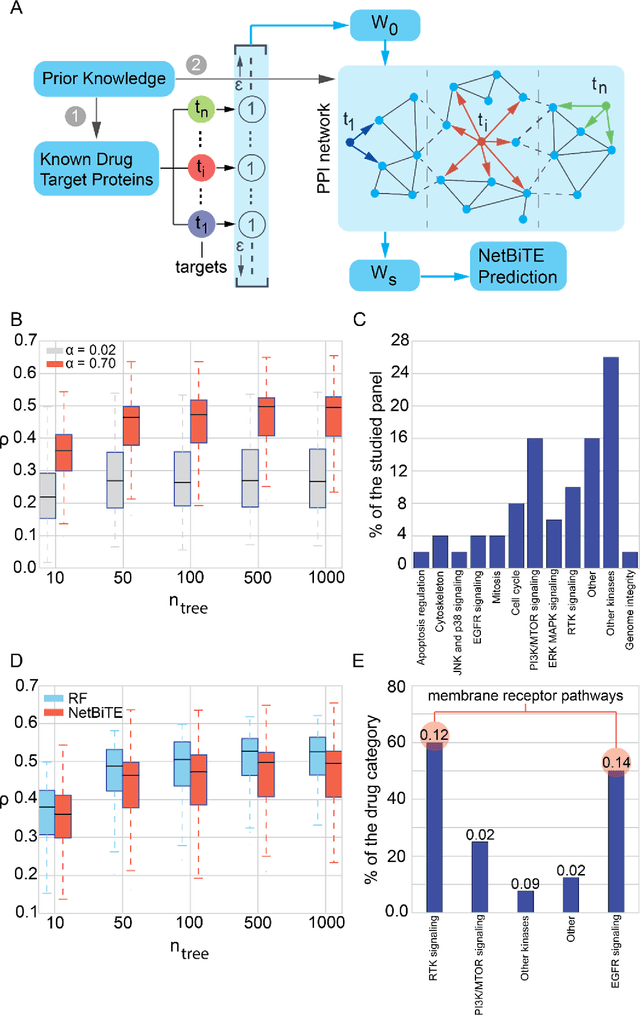
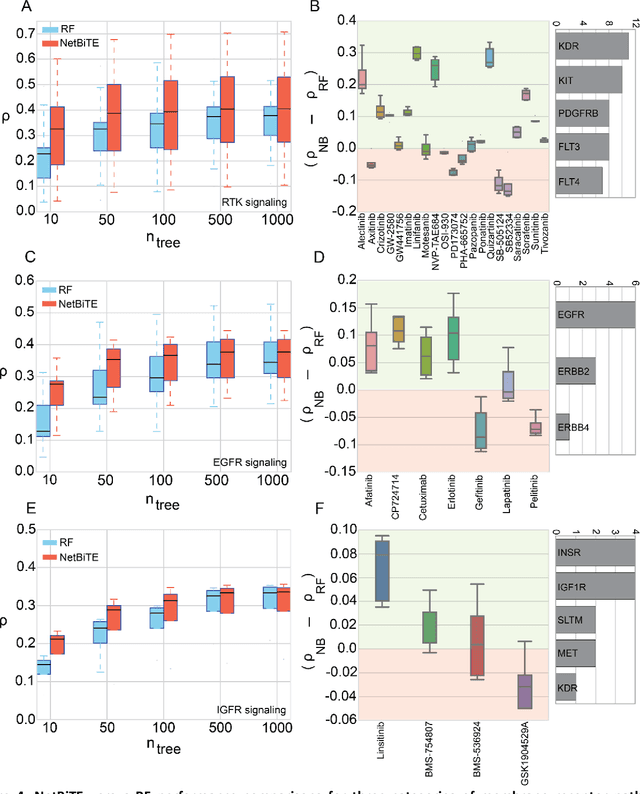
We present the Network-based Biased Tree Ensembles (NetBiTE) method for drug sensitivity prediction and drug sensitivity biomarker identification in cancer using a combination of prior knowledge and gene expression data. Our devised method consists of a biased tree ensemble that is built according to a probabilistic bias weight distribution. The bias weight distribution is obtained from the assignment of high weights to the drug targets and propagating the assigned weights over a protein-protein interaction network such as STRING. The propagation of weights, defines neighborhoods of influence around the drug targets and as such simulates the spread of perturbations within the cell, following drug administration. Using a synthetic dataset, we showcase how application of biased tree ensembles (BiTE) results in significant accuracy gains at a much lower computational cost compared to the unbiased random forests (RF) algorithm. We then apply NetBiTE to the Genomics of Drug Sensitivity in Cancer (GDSC) dataset and demonstrate that NetBiTE outperforms RF in predicting IC50 drug sensitivity, only for drugs that target membrane receptor pathways (MRPs): RTK, EGFR and IGFR signaling pathways. We propose based on the NetBiTE results, that for drugs that inhibit MRPs, the expression of target genes prior to drug administration is a biomarker for IC50 drug sensitivity following drug administration. We further verify and reinforce this proposition through control studies on, PI3K/MTOR signaling pathway inhibitors, a drug category that does not target MRPs, and through assignment of dummy targets to MRP inhibiting drugs and investigating the variation in NetBiTE accuracy.