 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edge"Stupid robot, I want to speak to a human!" User Frustration Detection in Task-Oriented Dialog Systems
Nov 26, 2024



Detecting user frustration in modern-day task-oriented dialog (TOD) systems is imperative for maintaining overall user satisfaction, engagement, and retention. However, most recent research is focused on sentiment and emotion detection in academic settings, thus failing to fully encapsulate implications of real-world user data. To mitigate this gap, in this work, we focus on user frustration in a deployed TOD system, assessing the feasibility of out-of-the-box solutions for user frustration detection. Specifically, we compare the performance of our deployed keyword-based approach, open-source approaches to sentiment analysis, dialog breakdown detection methods, and emerging in-context learning LLM-based detection. Our analysis highlights the limitations of open-source methods for real-world frustration detection, while demonstrating the superior performance of the LLM-based approach, achieving a 16\% relative improvement in F1 score on an internal benchmark. Finally, we analyze advantages and limitations of our methods and provide an insight into user frustration detection task for industry practitioners.
Reliable LLM-based User Simulator for Task-Oriented Dialogue Systems
Feb 20, 2024



In the realm of dialogue systems, user simulation techniques have emerged as a game-changer, redefining the evaluation and enhancement of task-oriented dialogue (TOD) systems. These methods are crucial for replicating real user interactions, enabling applications like synthetic data augmentation, error detection, and robust evaluation. However, existing approaches often rely on rigid rule-based methods or on annotated data. This paper introduces DAUS, a Domain-Aware User Simulator. Leveraging large language models, we fine-tune DAUS on real examples of task-oriented dialogues. Results on two relevant benchmarks showcase significant improvements in terms of user goal fulfillment. Notably, we have observed that fine-tuning enhances the simulator's coherence with user goals, effectively mitigating hallucinations -- a major source of inconsistencies in simulator responses.
In-Context Learning User Simulators for Task-Oriented Dialog Systems
Jun 01, 2023



This paper presents a novel application of large language models in user simulation for task-oriented dialog systems, specifically focusing on an in-context learning approach. By harnessing the power of these models, the proposed approach generates diverse utterances based on user goals and limited dialog examples. Unlike traditional simulators, this method eliminates the need for labor-intensive rule definition or extensive annotated data, making it more efficient and accessible. Additionally, an error analysis of the interaction between the user simulator and dialog system uncovers common mistakes, providing valuable insights into areas that require improvement. Our implementation is available at https://github.com/telepathylabsai/prompt-based-user-simulator.
Network-based Biased Tree Ensembles for Drug Sensitivity Prediction and Drug Sensitivity Biomarker Identification in Cancer
Aug 18, 2018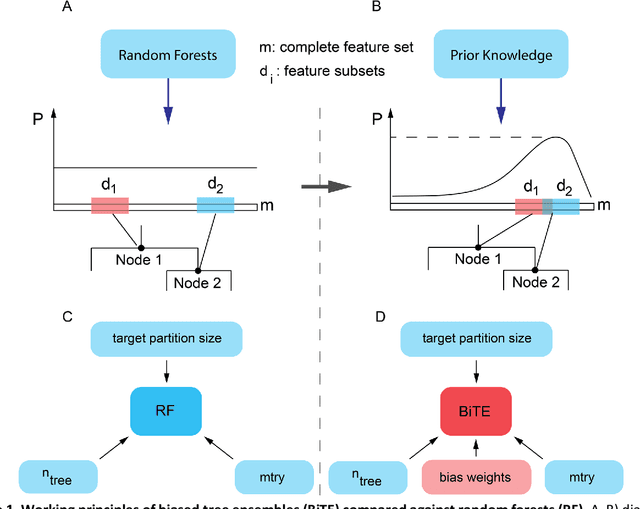
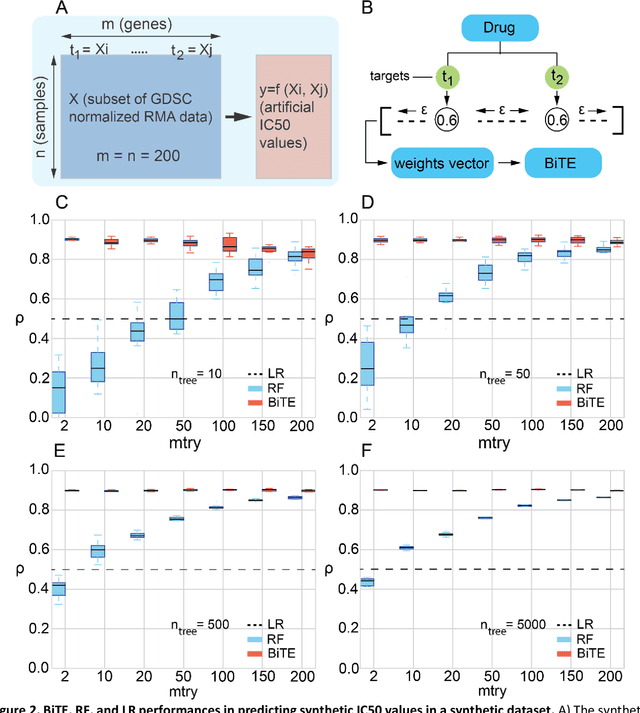
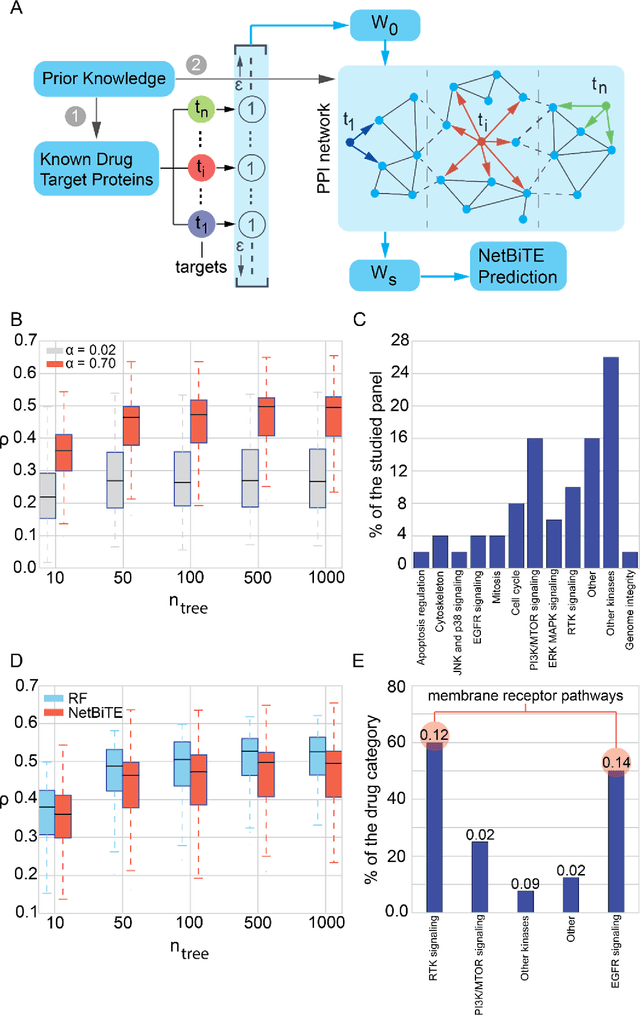
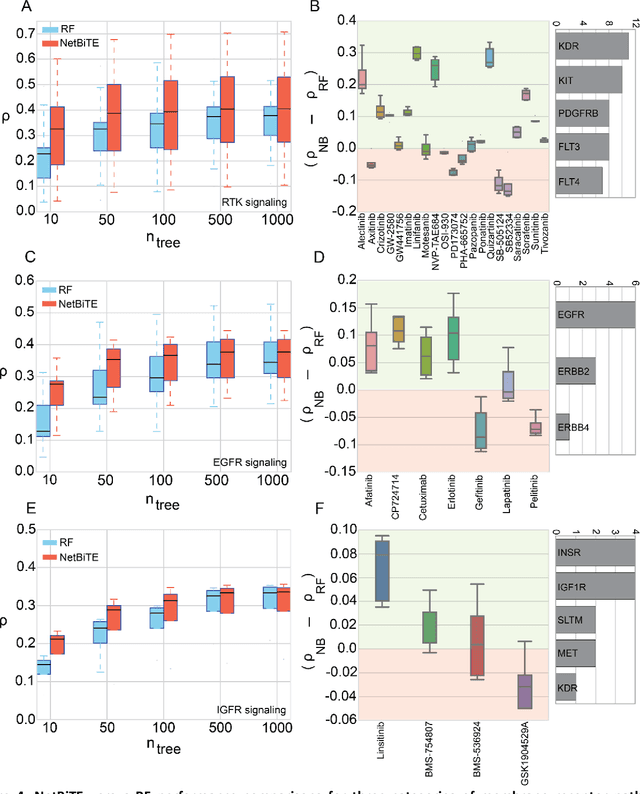
We present the Network-based Biased Tree Ensembles (NetBiTE) method for drug sensitivity prediction and drug sensitivity biomarker identification in cancer using a combination of prior knowledge and gene expression data. Our devised method consists of a biased tree ensemble that is built according to a probabilistic bias weight distribution. The bias weight distribution is obtained from the assignment of high weights to the drug targets and propagating the assigned weights over a protein-protein interaction network such as STRING. The propagation of weights, defines neighborhoods of influence around the drug targets and as such simulates the spread of perturbations within the cell, following drug administration. Using a synthetic dataset, we showcase how application of biased tree ensembles (BiTE) results in significant accuracy gains at a much lower computational cost compared to the unbiased random forests (RF) algorithm. We then apply NetBiTE to the Genomics of Drug Sensitivity in Cancer (GDSC) dataset and demonstrate that NetBiTE outperforms RF in predicting IC50 drug sensitivity, only for drugs that target membrane receptor pathways (MRPs): RTK, EGFR and IGFR signaling pathways. We propose based on the NetBiTE results, that for drugs that inhibit MRPs, the expression of target genes prior to drug administration is a biomarker for IC50 drug sensitivity following drug administration. We further verify and reinforce this proposition through control studies on, PI3K/MTOR signaling pathway inhibitors, a drug category that does not target MRPs, and through assignment of dummy targets to MRP inhibiting drugs and investigating the variation in NetBiTE accuracy.
PIMKL: Pathway Induced Multiple Kernel Learning
Jul 05, 2018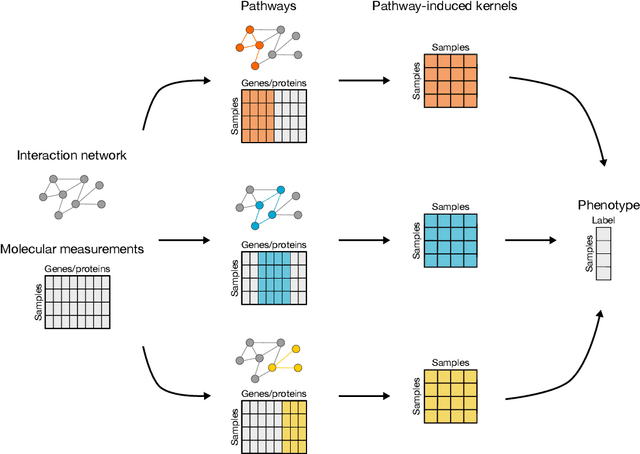
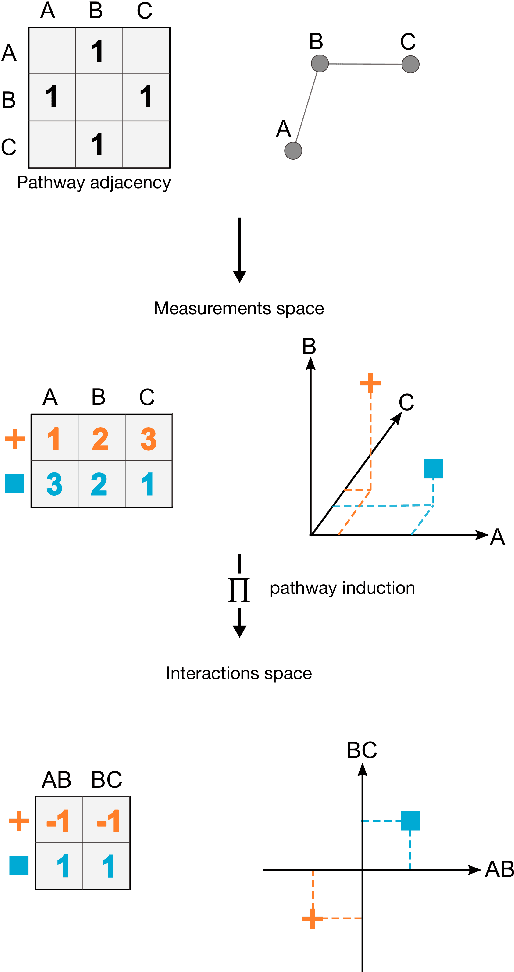
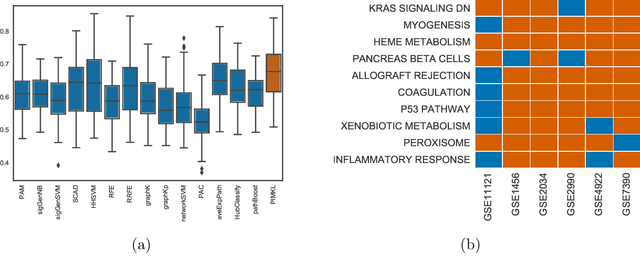
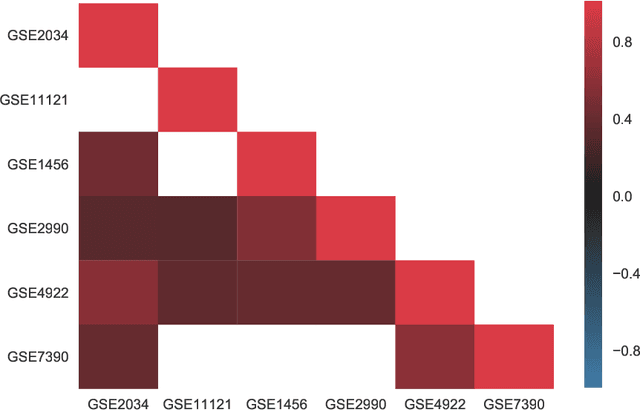
Reliable identification of molecular biomarkers is essential for accurate patient stratification. While state-of-the-art machine learning approaches for sample classification continue to push boundaries in terms of performance, most of these methods are not able to integrate different data types and lack generalization power, limiting their application in a clinical setting. Furthermore, many methods behave as black boxes, and we have very little understanding about the mechanisms that lead to the prediction. While opaqueness concerning machine behaviour might not be a problem in deterministic domains, in health care, providing explanations about the molecular factors and phenotypes that are driving the classification is crucial to build trust in the performance of the predictive system. We propose Pathway Induced Multiple Kernel Learning (PIMKL), a novel methodology to reliably classify samples that can also help gain insights into the molecular mechanisms that underlie the classification. PIMKL exploits prior knowledge in the form of a molecular interaction network and annotated gene sets, by optimizing a mixture of pathway-induced kernels using a Multiple Kernel Learning (MKL) algorithm, an approach that has demonstrated excellent performance in different machine learning applications. After optimizing the combination of kernels for prediction of a specific phenotype, the model provides a stable molecular signature that can be interpreted in the light of the ingested prior knowledge and that can be used in transfer learning tasks.