 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFRED: The Florence RGB-Event Drone Dataset
Jun 05, 2025Small, fast, and lightweight drones present significant challenges for traditional RGB cameras due to their limitations in capturing fast-moving objects, especially under challenging lighting conditions. Event cameras offer an ideal solution, providing high temporal definition and dynamic range, yet existing benchmarks often lack fine temporal resolution or drone-specific motion patterns, hindering progress in these areas. This paper introduces the Florence RGB-Event Drone dataset (FRED), a novel multimodal dataset specifically designed for drone detection, tracking, and trajectory forecasting, combining RGB video and event streams. FRED features more than 7 hours of densely annotated drone trajectories, using 5 different drone models and including challenging scenarios such as rain and adverse lighting conditions. We provide detailed evaluation protocols and standard metrics for each task, facilitating reproducible benchmarking. The authors hope FRED will advance research in high-speed drone perception and multimodal spatiotemporal understanding.
Automatic Labels are as Effective as Manual Labels in Biomedical Images Classification with Deep Learning
Jun 20, 2024The increasing availability of biomedical data is helping to design more robust deep learning (DL) algorithms to analyze biomedical samples. Currently, one of the main limitations to train DL algorithms to perform a specific task is the need for medical experts to label data. Automatic methods to label data exist, however automatic labels can be noisy and it is not completely clear when automatic labels can be adopted to train DL models. This paper aims to investigate under which circumstances automatic labels can be adopted to train a DL model on the classification of Whole Slide Images (WSI). The analysis involves multiple architectures, such as Convolutional Neural Networks (CNN) and Vision Transformer (ViT), and over 10000 WSIs, collected from three use cases: celiac disease, lung cancer and colon cancer, which one including respectively binary, multiclass and multilabel data. The results allow identifying 10% as the percentage of noisy labels that lead to train competitive models for the classification of WSIs. Therefore, an algorithm generating automatic labels needs to fit this criterion to be adopted. The application of the Semantic Knowledge Extractor Tool (SKET) algorithm to generate automatic labels leads to performance comparable to the one obtained with manual labels, since it generates a percentage of noisy labels between 2-5%. Automatic labels are as effective as manual ones, reaching solid performance comparable to the one obtained training models with manual labels.
RegWSI: Whole Slide Image Registration using Combined Deep Feature- and Intensity-Based Methods: Winner of the ACROBAT 2023 Challenge
Apr 26, 2024



The automatic registration of differently stained whole slide images (WSIs) is crucial for improving diagnosis and prognosis by fusing complementary information emerging from different visible structures. It is also useful to quickly transfer annotations between consecutive or restained slides, thus significantly reducing the annotation time and associated costs. Nevertheless, the slide preparation is different for each stain and the tissue undergoes complex and large deformations. Therefore, a robust, efficient, and accurate registration method is highly desired by the scientific community and hospitals specializing in digital pathology. We propose a two-step hybrid method consisting of (i) deep learning- and feature-based initial alignment algorithm, and (ii) intensity-based nonrigid registration using the instance optimization. The proposed method does not require any fine-tuning to a particular dataset and can be used directly for any desired tissue type and stain. The method scored 1st place in the ACROBAT 2023 challenge. We evaluated using three open datasets: (i) ANHIR, (ii) ACROBAT, and (iii) HyReCo, and performed several ablation studies concerning the resolution used for registration and the initial alignment robustness and stability. The method achieves the most accurate results for the ACROBAT dataset, the cell-level registration accuracy for the restained slides from the HyReCo dataset, and is among the best methods evaluated on the ANHIR dataset. The method does not require any fine-tuning to a new datasets and can be used out-of-the-box for other types of microscopic images. The method is incorporated into the DeeperHistReg framework, allowing others to directly use it to register, transform, and save the WSIs at any desired pyramid level. The proposed method is a significant contribution to the WSI registration, thus advancing the field of digital pathology.
DeeperHistReg: Robust Whole Slide Images Registration Framework
Apr 19, 2024DeeperHistReg is a software framework dedicated to registering whole slide images (WSIs) acquired using multiple stains. It allows one to perform the preprocessing, initial alignment, and nonrigid registration of WSIs acquired using multiple stains (e.g. hematoxylin \& eosin, immunochemistry). The framework implements several state-of-the-art registration algorithms and provides an interface to operate on arbitrary resolution of the WSIs (up to 200k x 200k). The framework is extensible and new algorithms can be easily integrated by other researchers. The framework is available both as a PyPI package and as a Docker container.
The ACROBAT 2022 Challenge: Automatic Registration Of Breast Cancer Tissue
May 29, 2023The alignment of tissue between histopathological whole-slide-images (WSI) is crucial for research and clinical applications. Advances in computing, deep learning, and availability of large WSI datasets have revolutionised WSI analysis. Therefore, the current state-of-the-art in WSI registration is unclear. To address this, we conducted the ACROBAT challenge, based on the largest WSI registration dataset to date, including 4,212 WSIs from 1,152 breast cancer patients. The challenge objective was to align WSIs of tissue that was stained with routine diagnostic immunohistochemistry to its H&E-stained counterpart. We compare the performance of eight WSI registration algorithms, including an investigation of the impact of different WSI properties and clinical covariates. We find that conceptually distinct WSI registration methods can lead to highly accurate registration performances and identify covariates that impact performances across methods. These results establish the current state-of-the-art in WSI registration and guide researchers in selecting and developing methods.
Attention-based Interpretable Regression of Gene Expression in Histology
Aug 29, 2022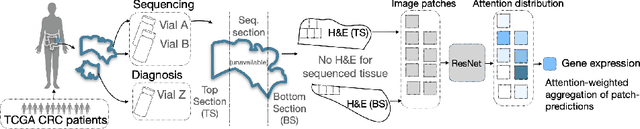
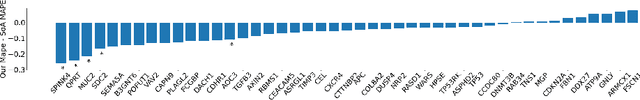


Interpretability of deep learning is widely used to evaluate the reliability of medical imaging models and reduce the risks of inaccurate patient recommendations. For models exceeding human performance, e.g. predicting RNA structure from microscopy images, interpretable modelling can be further used to uncover highly non-trivial patterns which are otherwise imperceptible to the human eye. We show that interpretability can reveal connections between the microscopic appearance of cancer tissue and its gene expression profiling. While exhaustive profiling of all genes from the histology images is still challenging, we estimate the expression values of a well-known subset of genes that is indicative of cancer molecular subtype, survival, and treatment response in colorectal cancer. Our approach successfully identifies meaningful information from the image slides, highlighting hotspots of high gene expression. Our method can help characterise how gene expression shapes tissue morphology and this may be beneficial for patient stratification in the pathology unit. The code is available on GitHub.