 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeValidation of an AI-based end-to-end model for prostate pathology using long-term archived routine samples
May 04, 2026Artificial intelligence (AI) is becoming a clinical tool for prostate pathology, but generalization across variations in sample preparation and preservation over prolonged time periods remains poorly understood. We evaluated GleasonAI, an end-to-end attention-based multiple instance learning model, on an independent validation cohort comprising 10,366 biopsy cores from 1,028 patients across 14 Swedish regions, using archival diagnostic specimens from the ProMort cohorts collected between 1998-2015. The model achieved an overall quadratic-weighted kappa of 0.86 for core-level ISUP grading, comparable to several experienced pathologists and consistent across geographic regions. Notably, performance remained stable across the 17-year collection period, demonstrating robustness to time-related variation in archival material, a property not consistently observed with foundation model-based approaches, with exploratory analysis demonstrating a significant prognostic gradient across AI-assigned grade groups for prostate cancer-specific mortality. These findings support the generalizability of the AI grading model and demonstrate the potential of pathology archives as a large-scale resource for AI development, validation, and retrospective prognostic research.
AI-based Prediction of Biochemical Recurrence from Biopsy and Prostatectomy Samples
Jan 28, 2026Biochemical recurrence (BCR) after radical prostatectomy (RP) is a surrogate marker for aggressive prostate cancer with adverse outcomes, yet current prognostic tools remain imprecise. We trained an AI-based model on diagnostic prostate biopsy slides from the STHLM3 cohort (n = 676) to predict patient-specific risk of BCR, using foundation models and attention-based multiple instance learning. Generalizability was assessed across three external RP cohorts: LEOPARD (n = 508), CHIMERA (n = 95), and TCGA-PRAD (n = 379). The image-based approach achieved 5-year time-dependent AUCs of 0.64, 0.70, and 0.70, respectively. Integrating clinical variables added complementary prognostic value and enabled statistically significant risk stratification. Compared with guideline-based CAPRA-S, AI incrementally improved postoperative prognostication. These findings suggest biopsy-trained histopathology AI can generalize across specimen types to support preoperative and postoperative decision making, but the added value of AI-based multimodal approaches over simpler predictive models should be critically scrutinized in further studies.
Validation of Diagnostic Artificial Intelligence Models for Prostate Pathology in a Middle Eastern Cohort
Dec 19, 2025Background: Artificial intelligence (AI) is improving the efficiency and accuracy of cancer diagnostics. The performance of pathology AI systems has been almost exclusively evaluated on European and US cohorts from large centers. For global AI adoption in pathology, validation studies on currently under-represented populations - where the potential gains from AI support may also be greatest - are needed. We present the first study with an external validation cohort from the Middle East, focusing on AI-based diagnosis and Gleason grading of prostate cancer. Methods: We collected and digitised 339 prostate biopsy specimens from the Kurdistan region, Iraq, representing a consecutive series of 185 patients spanning the period 2013-2024. We evaluated a task-specific end-to-end AI model and two foundation models in terms of their concordance with pathologists and consistency across samples digitised on three scanner models (Hamamatsu, Leica, and Grundium). Findings: Grading concordance between AI and pathologists was similar to pathologist-pathologist concordance with Cohen's quadratically weighted kappa 0.801 vs. 0.799 (p=0.9824). Cross-scanner concordance was high (quadratically weighted kappa > 0.90) for all AI models and scanner pairs, including low-cost compact scanner. Interpretation: AI models demonstrated pathologist-level performance in prostate histopathology assessment. Compact scanners can provide a route for validation studies in non-digitalised settings and enable cost-effective adoption of AI in laboratories with limited sample volumes. This first openly available digital pathology dataset from the Middle East supports further research into globally equitable AI pathology. Funding: SciLifeLab and Wallenberg Data Driven Life Science Program, Instrumentarium Science Foundation, Karolinska Institutet Research Foundation.
Artificial Intelligence-Assisted Prostate Cancer Diagnosis for Reduced Use of Immunohistochemistry
Mar 31, 2025Prostate cancer diagnosis heavily relies on histopathological evaluation, which is subject to variability. While immunohistochemical staining (IHC) assists in distinguishing benign from malignant tissue, it involves increased work, higher costs, and diagnostic delays. Artificial intelligence (AI) presents a promising solution to reduce reliance on IHC by accurately classifying atypical glands and borderline morphologies in hematoxylin & eosin (H&E) stained tissue sections. In this study, we evaluated an AI model's ability to minimize IHC use without compromising diagnostic accuracy by retrospectively analyzing prostate core needle biopsies from routine diagnostics at three different pathology sites. These cohorts were composed exclusively of difficult cases where the diagnosing pathologists required IHC to finalize the diagnosis. The AI model demonstrated area under the curve values of 0.951-0.993 for detecting cancer in routine H&E-stained slides. Applying sensitivity-prioritized diagnostic thresholds reduced the need for IHC staining by 44.4%, 42.0%, and 20.7% in the three cohorts investigated, without a single false negative prediction. This AI model shows potential for optimizing IHC use, streamlining decision-making in prostate pathology, and alleviating resource burdens.
The impact of tissue detection on diagnostic artificial intelligence algorithms in digital pathology
Mar 29, 2025
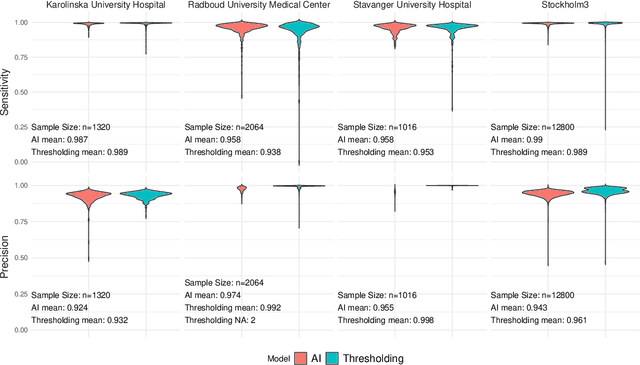


Tissue detection is a crucial first step in most digital pathology applications. Details of the segmentation algorithm are rarely reported, and there is a lack of studies investigating the downstream effects of a poor segmentation algorithm. Disregarding tissue detection quality could create a bottleneck for downstream performance and jeopardize patient safety if diagnostically relevant parts of the specimen are excluded from analysis in clinical applications. This study aims to determine whether performance of downstream tasks is sensitive to the tissue detection method, and to compare performance of classical and AI-based tissue detection. To this end, we trained an AI model for Gleason grading of prostate cancer in whole slide images (WSIs) using two different tissue detection algorithms: thresholding (classical) and UNet++ (AI). A total of 33,823 WSIs scanned on five digital pathology scanners were used to train the tissue detection AI model. The downstream Gleason grading algorithm was trained and tested using 70,524 WSIs from 13 clinical sites scanned on 13 different scanners. There was a decrease from 116 (0.43%) to 22 (0.08%) fully undetected tissue samples when switching from thresholding-based tissue detection to AI-based, suggesting an AI model may be more reliable than a classical model for avoiding total failures on slides with unusual appearance. On the slides where tissue could be detected by both algorithms, no significant difference in overall Gleason grading performance was observed. However, tissue detection dependent clinically significant variations in AI grading were observed in 3.5% of malignant slides, highlighting the importance of robust tissue detection for optimal clinical performance of diagnostic AI.
Foundation Models -- A Panacea for Artificial Intelligence in Pathology?
Feb 28, 2025
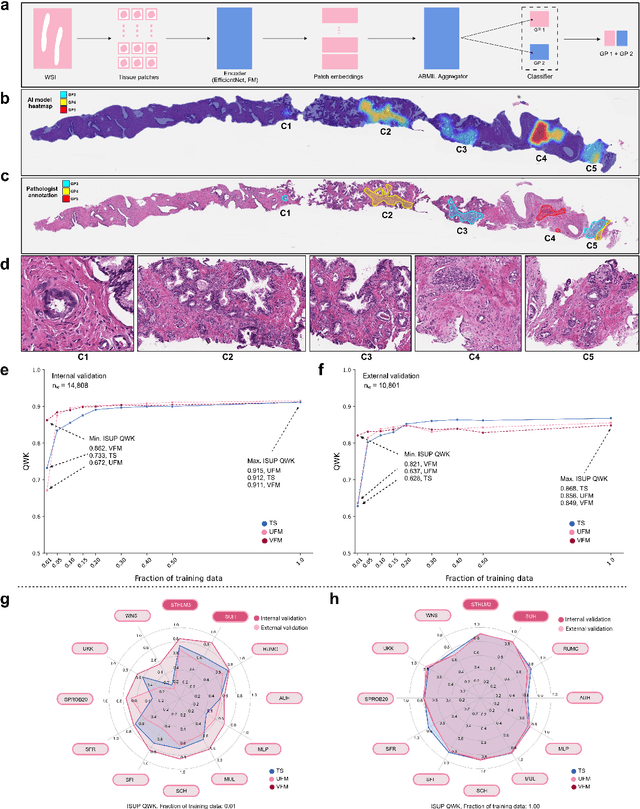

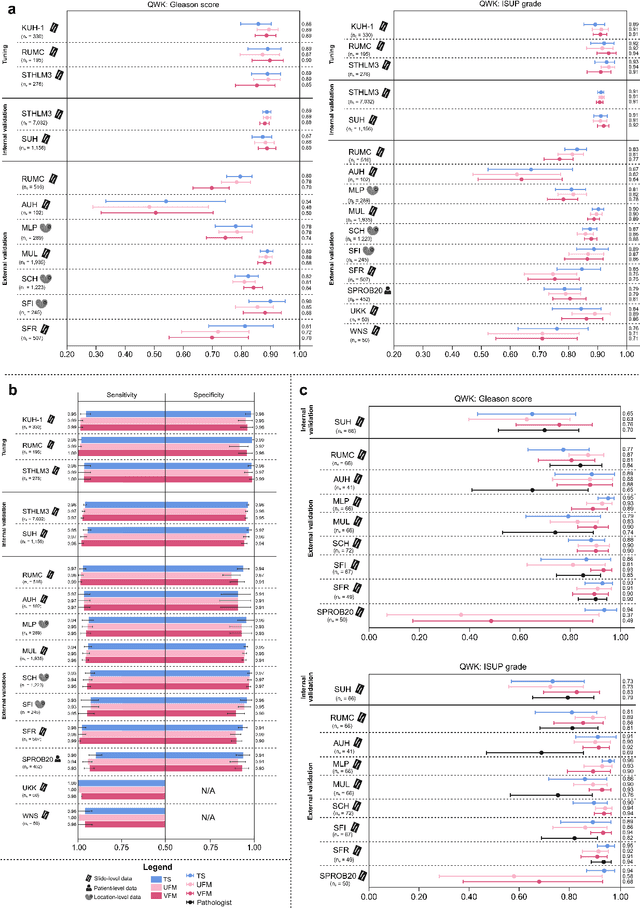
The role of artificial intelligence (AI) in pathology has evolved from aiding diagnostics to uncovering predictive morphological patterns in whole slide images (WSIs). Recently, foundation models (FMs) leveraging self-supervised pre-training have been widely advocated as a universal solution for diverse downstream tasks. However, open questions remain about their clinical applicability and generalization advantages over end-to-end learning using task-specific (TS) models. Here, we focused on AI with clinical-grade performance for prostate cancer diagnosis and Gleason grading. We present the largest validation of AI for this task, using over 100,000 core needle biopsies from 7,342 patients across 15 sites in 11 countries. We compared two FMs with a fully end-to-end TS model in a multiple instance learning framework. Our findings challenge assumptions that FMs universally outperform TS models. While FMs demonstrated utility in data-scarce scenarios, their performance converged with - and was in some cases surpassed by - TS models when sufficient labeled training data were available. Notably, extensive task-specific training markedly reduced clinically significant misgrading, misdiagnosis of challenging morphologies, and variability across different WSI scanners. Additionally, FMs used up to 35 times more energy than the TS model, raising concerns about their sustainability. Our results underscore that while FMs offer clear advantages for rapid prototyping and research, their role as a universal solution for clinically applicable medical AI remains uncertain. For high-stakes clinical applications, rigorous validation and consideration of task-specific training remain critically important. We advocate for integrating the strengths of FMs and end-to-end learning to achieve robust and resource-efficient AI pathology solutions fit for clinical use.
Physical Color Calibration of Digital Pathology Scanners for Robust Artificial Intelligence Assisted Cancer Diagnosis
Jul 07, 2023



The potential of artificial intelligence (AI) in digital pathology is limited by technical inconsistencies in the production of whole slide images (WSIs), leading to degraded AI performance and posing a challenge for widespread clinical application as fine-tuning algorithms for each new site is impractical. Changes in the imaging workflow can also lead to compromised diagnoses and patient safety risks. We evaluated whether physical color calibration of scanners can standardize WSI appearance and enable robust AI performance. We employed a color calibration slide in four different laboratories and evaluated its impact on the performance of an AI system for prostate cancer diagnosis on 1,161 WSIs. Color standardization resulted in consistently improved AI model calibration and significant improvements in Gleason grading performance. The study demonstrates that physical color calibration provides a potential solution to the variation introduced by different scanners, making AI-based cancer diagnostics more reliable and applicable in clinical settings.
The ACROBAT 2022 Challenge: Automatic Registration Of Breast Cancer Tissue
May 29, 2023The alignment of tissue between histopathological whole-slide-images (WSI) is crucial for research and clinical applications. Advances in computing, deep learning, and availability of large WSI datasets have revolutionised WSI analysis. Therefore, the current state-of-the-art in WSI registration is unclear. To address this, we conducted the ACROBAT challenge, based on the largest WSI registration dataset to date, including 4,212 WSIs from 1,152 breast cancer patients. The challenge objective was to align WSIs of tissue that was stained with routine diagnostic immunohistochemistry to its H&E-stained counterpart. We compare the performance of eight WSI registration algorithms, including an investigation of the impact of different WSI properties and clinical covariates. We find that conceptually distinct WSI registration methods can lead to highly accurate registration performances and identify covariates that impact performances across methods. These results establish the current state-of-the-art in WSI registration and guide researchers in selecting and developing methods.
ACROBAT -- a multi-stain breast cancer histological whole-slide-image data set from routine diagnostics for computational pathology
Nov 24, 2022The analysis of FFPE tissue sections stained with haematoxylin and eosin (H&E) or immunohistochemistry (IHC) is an essential part of the pathologic assessment of surgically resected breast cancer specimens. IHC staining has been broadly adopted into diagnostic guidelines and routine workflows to manually assess status and scoring of several established biomarkers, including ER, PGR, HER2 and KI67. However, this is a task that can also be facilitated by computational pathology image analysis methods. The research in computational pathology has recently made numerous substantial advances, often based on publicly available whole slide image (WSI) data sets. However, the field is still considerably limited by the sparsity of public data sets. In particular, there are no large, high quality publicly available data sets with WSIs of matching IHC and H&E-stained tissue sections. Here, we publish the currently largest publicly available data set of WSIs of tissue sections from surgical resection specimens from female primary breast cancer patients with matched WSIs of corresponding H&E and IHC-stained tissue, consisting of 4,212 WSIs from 1,153 patients. The primary purpose of the data set was to facilitate the ACROBAT WSI registration challenge, aiming at accurately aligning H&E and IHC images. For research in the area of image registration, automatic quantitative feedback on registration algorithm performance remains available through the ACROBAT challenge website, based on more than 37,000 manually annotated landmark pairs from 13 annotators. Beyond registration, this data set has the potential to enable many different avenues of computational pathology research, including stain-guided learning, virtual staining, unsupervised pre-training, artefact detection and stain-independent models.
Transcriptome-wide prediction of prostate cancer gene expression from histopathology images using co-expression based convolutional neural networks
Apr 19, 2021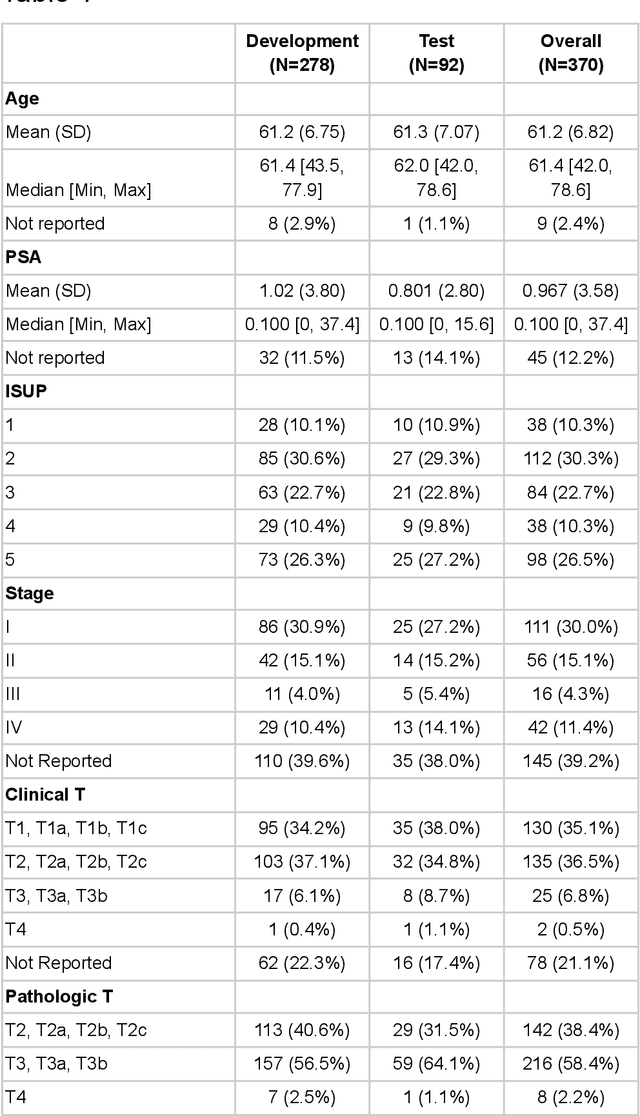
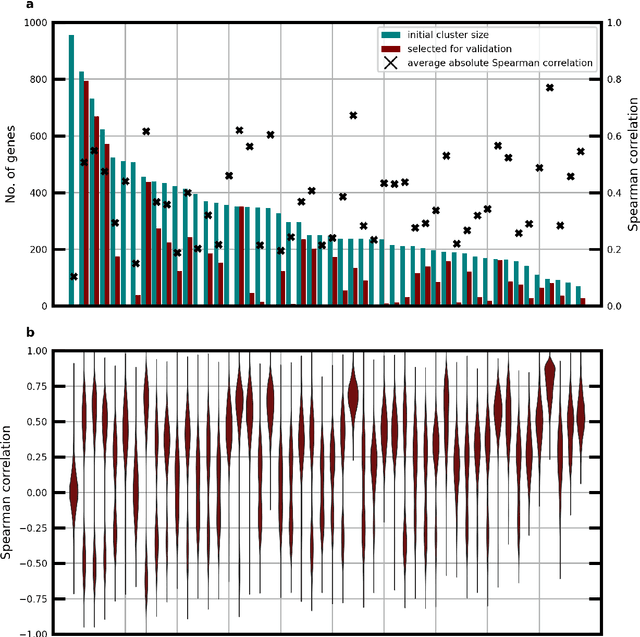
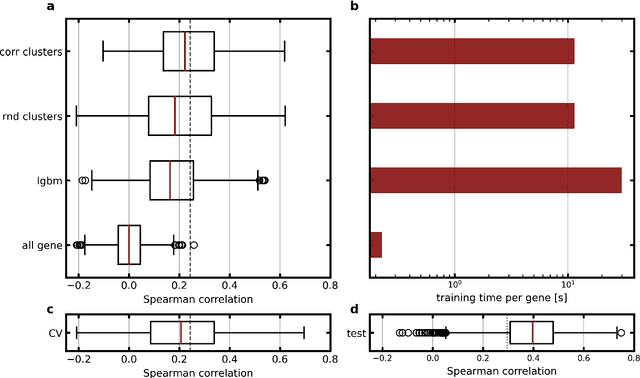
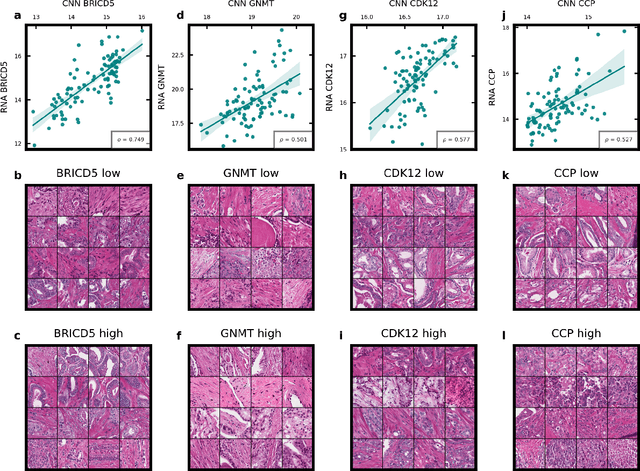
Molecular phenotyping by gene expression profiling is common in contemporary cancer research and in molecular diagnostics. However, molecular profiling remains costly and resource intense to implement, and is just starting to be introduced into clinical diagnostics. Molecular changes, including genetic alterations and gene expression changes, occuring in tumors cause morphological changes in tissue, which can be observed on the microscopic level. The relationship between morphological patterns and some of the molecular phenotypes can be exploited to predict molecular phenotypes directly from routine haematoxylin and eosin (H&E) stained whole slide images (WSIs) using deep convolutional neural networks (CNNs). In this study, we propose a new, computationally efficient approach for disease specific modelling of relationships between morphology and gene expression, and we conducted the first transcriptome-wide analysis in prostate cancer, using CNNs to predict bulk RNA-sequencing estimates from WSIs of H&E stained tissue. The work is based on the TCGA PRAD study and includes both WSIs and RNA-seq data for 370 patients. Out of 15586 protein coding and sufficiently frequently expressed transcripts, 6618 had predicted expression significantly associated with RNA-seq estimates (FDR-adjusted p-value < 1*10-4) in a cross-validation. 5419 (81.9%) of these were subsequently validated in a held-out test set. We also demonstrate the ability to predict a prostate cancer specific cell cycle progression score directly from WSIs. These findings suggest that contemporary computer vision models offer an inexpensive and scalable solution for prediction of gene expression phenotypes directly from WSIs, providing opportunity for cost-effective large-scale research studies and molecular diagnostics.