 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScanner-Induced Domain Shifts Undermine the Robustness of Pathology Foundation Models
Jan 07, 2026Pathology foundation models (PFMs) have become central to computational pathology, aiming to offer general encoders for feature extraction from whole-slide images (WSIs). Despite strong benchmark performance, PFM robustness to real-world technical domain shifts, such as variability from whole-slide scanner devices, remains poorly understood. We systematically evaluated the robustness of 14 PFMs to scanner-induced variability, including state-of-the-art models, earlier self-supervised models, and a baseline trained on natural images. Using a multiscanner dataset of 384 breast cancer WSIs scanned on five devices, we isolated scanner effects independently from biological and laboratory confounders. Robustness is assessed via complementary unsupervised embedding analyses and a set of clinicopathological supervised prediction tasks. Our results demonstrate that current PFMs are not invariant to scanner-induced domain shifts. Most models encode pronounced scanner-specific variability in their embedding spaces. While AUC often remains stable, this masks a critical failure mode: scanner variability systematically alters the embedding space and impacts calibration of downstream model predictions, resulting in scanner-dependent bias that can impact reliability in clinical use cases. We further show that robustness is not a simple function of training data scale, model size, or model recency. None of the models provided reliable robustness against scanner-induced variability. While the models trained on the most diverse data, here represented by vision-language models, appear to have an advantage with respect to robustness, they underperformed on downstream supervised tasks. We conclude that development and evaluation of PFMs requires moving beyond accuracy-centric benchmarks toward explicit evaluation and optimisation of embedding stability and calibration under realistic acquisition variability.
Evaluating Computational Pathology Foundation Models for Prostate Cancer Grading under Distribution Shifts
Oct 09, 2024



Foundation models have recently become a popular research direction within computational pathology. They are intended to be general-purpose feature extractors, promising to achieve good performance on a range of downstream tasks. Real-world pathology image data does however exhibit considerable variability. Foundation models should be robust to these variations and other distribution shifts which might be encountered in practice. We evaluate two computational pathology foundation models: UNI (trained on more than 100,000 whole-slide images) and CONCH (trained on more than 1.1 million image-caption pairs), by utilizing them as feature extractors within prostate cancer grading models. We find that while UNI and CONCH perform well relative to baselines, the absolute performance can still be far from satisfactory in certain settings. The fact that foundation models have been trained on large and varied datasets does not guarantee that downstream models always will be robust to common distribution shifts.
Evaluating Deep Regression Models for WSI-Based Gene-Expression Prediction
Oct 01, 2024Prediction of mRNA gene-expression profiles directly from routine whole-slide images (WSIs) using deep learning models could potentially offer cost-effective and widely accessible molecular phenotyping. While such WSI-based gene-expression prediction models have recently emerged within computational pathology, the high-dimensional nature of the corresponding regression problem offers numerous design choices which remain to be analyzed in detail. This study provides recommendations on how deep regression models should be trained for WSI-based gene-expression prediction. For example, we conclude that training a single model to simultaneously regress all 20530 genes is a computationally efficient yet very strong baseline.
Deep Blur Multi-Model -- a strategy to mitigate the impact of image blur on deep learning model performance in histopathology image analysis
May 15, 2024AI-based analysis of histopathology whole slide images (WSIs) is central in computational pathology. However, image quality can impact model performance. Here, we investigate to what extent unsharp areas of WSIs impact deep convolutional neural network classification performance. We propose a multi-model approach, i.e. DeepBlurMM, to alleviate the impact of unsharp image areas and improve the model performance. DeepBlurMM uses the sigma cut-offs to determine the most suitable model for predicting tiles with various levels of blurring within a single WSI, where sigma is the standard deviation of the Gaussian distribution. Specifically, the cut-offs categorise the tiles into sharp or slight blur, moderate blur, and high blur. Each blur level has a corresponding model to be selected for tile-level predictions. Throughout the simulation study, we demonstrated the application of DeepBlurMM in a binary classification task for breast cancer Nottingham Histological Grade 1 vs 3. Performance, evaluated over 5-fold cross-validation, showed that DeepBlurMM outperformed the base model under moderate blur and mixed blur conditions. Unsharp image tiles (local blurriness) at prediction time reduced model performance. The proposed multi-model approach improved performance under some conditions, with the potential to improve quality in both research and clinical applications.
WEEP: A method for spatial interpretation of weakly supervised CNN models in computational pathology
Apr 08, 2024Deep learning enables the modelling of high-resolution histopathology whole-slide images (WSI). Weakly supervised learning of tile-level data is typically applied for tasks where labels only exist on the patient or WSI level (e.g. patient outcomes or histological grading). In this context, there is a need for improved spatial interpretability of predictions from such models. We propose a novel method, Wsi rEgion sElection aPproach (WEEP), for model interpretation. It provides a principled yet straightforward way to establish the spatial area of WSI required for assigning a particular prediction label. We demonstrate WEEP on a binary classification task in the area of breast cancer computational pathology. WEEP is easy to implement, is directly connected to the model-based decision process, and offers information relevant to both research and diagnostic applications.
Physical Color Calibration of Digital Pathology Scanners for Robust Artificial Intelligence Assisted Cancer Diagnosis
Jul 07, 2023



The potential of artificial intelligence (AI) in digital pathology is limited by technical inconsistencies in the production of whole slide images (WSIs), leading to degraded AI performance and posing a challenge for widespread clinical application as fine-tuning algorithms for each new site is impractical. Changes in the imaging workflow can also lead to compromised diagnoses and patient safety risks. We evaluated whether physical color calibration of scanners can standardize WSI appearance and enable robust AI performance. We employed a color calibration slide in four different laboratories and evaluated its impact on the performance of an AI system for prostate cancer diagnosis on 1,161 WSIs. Color standardization resulted in consistently improved AI model calibration and significant improvements in Gleason grading performance. The study demonstrates that physical color calibration provides a potential solution to the variation introduced by different scanners, making AI-based cancer diagnostics more reliable and applicable in clinical settings.
The ACROBAT 2022 Challenge: Automatic Registration Of Breast Cancer Tissue
May 29, 2023The alignment of tissue between histopathological whole-slide-images (WSI) is crucial for research and clinical applications. Advances in computing, deep learning, and availability of large WSI datasets have revolutionised WSI analysis. Therefore, the current state-of-the-art in WSI registration is unclear. To address this, we conducted the ACROBAT challenge, based on the largest WSI registration dataset to date, including 4,212 WSIs from 1,152 breast cancer patients. The challenge objective was to align WSIs of tissue that was stained with routine diagnostic immunohistochemistry to its H&E-stained counterpart. We compare the performance of eight WSI registration algorithms, including an investigation of the impact of different WSI properties and clinical covariates. We find that conceptually distinct WSI registration methods can lead to highly accurate registration performances and identify covariates that impact performances across methods. These results establish the current state-of-the-art in WSI registration and guide researchers in selecting and developing methods.
Increasing the usefulness of already existing annotations through WSI registration
Mar 12, 2023Computational pathology methods have the potential to improve access to precision medicine, as well as the reproducibility and accuracy of pathological diagnoses. Particularly the analysis of whole-slide-images (WSIs) of immunohistochemically (IHC) stained tissue sections could benefit from computational pathology methods. However, scoring biomarkers such as KI67 in IHC WSIs often necessitates the detection of areas of invasive cancer. Training cancer detection models often requires annotations, which is time-consuming and therefore costly. Currently, cancer regions are typically annotated in WSIs of haematoxylin and eosin (H&E) stained tissue sections. In this study, we investigate the possibility to register annotations that were made in H&E WSIs to their IHC counterparts. Two pathologists annotated regions of invasive cancer in WSIs of 272 breast cancer cases. For each case, a matched H&E and KI67 WSI are available, resulting in 544 WSIs with invasive cancer annotations. We find that cancer detection CNNs that were trained with annotations registered from the H&E to the KI67 WSIs only differ slightly in calibration but not in performance compared to cancer detection models trained on annotations made directly in the KI67 WSIs in a test set consisting of 54 cases. The mean slide-level AUROC is 0.974 [0.964, 0.982] for models trained with the KI67 annotations and 0.974 [0.965, 0.982] for models trained using registered annotations. This indicates that WSI registration has the potential to reduce the need for IHC-specific annotations. This could significantly increase the usefulness of already existing annotations.
ACROBAT -- a multi-stain breast cancer histological whole-slide-image data set from routine diagnostics for computational pathology
Nov 24, 2022The analysis of FFPE tissue sections stained with haematoxylin and eosin (H&E) or immunohistochemistry (IHC) is an essential part of the pathologic assessment of surgically resected breast cancer specimens. IHC staining has been broadly adopted into diagnostic guidelines and routine workflows to manually assess status and scoring of several established biomarkers, including ER, PGR, HER2 and KI67. However, this is a task that can also be facilitated by computational pathology image analysis methods. The research in computational pathology has recently made numerous substantial advances, often based on publicly available whole slide image (WSI) data sets. However, the field is still considerably limited by the sparsity of public data sets. In particular, there are no large, high quality publicly available data sets with WSIs of matching IHC and H&E-stained tissue sections. Here, we publish the currently largest publicly available data set of WSIs of tissue sections from surgical resection specimens from female primary breast cancer patients with matched WSIs of corresponding H&E and IHC-stained tissue, consisting of 4,212 WSIs from 1,153 patients. The primary purpose of the data set was to facilitate the ACROBAT WSI registration challenge, aiming at accurately aligning H&E and IHC images. For research in the area of image registration, automatic quantitative feedback on registration algorithm performance remains available through the ACROBAT challenge website, based on more than 37,000 manually annotated landmark pairs from 13 annotators. Beyond registration, this data set has the potential to enable many different avenues of computational pathology research, including stain-guided learning, virtual staining, unsupervised pre-training, artefact detection and stain-independent models.
Using deep learning to detect patients at risk for prostate cancer despite benign biopsies
Jul 31, 2021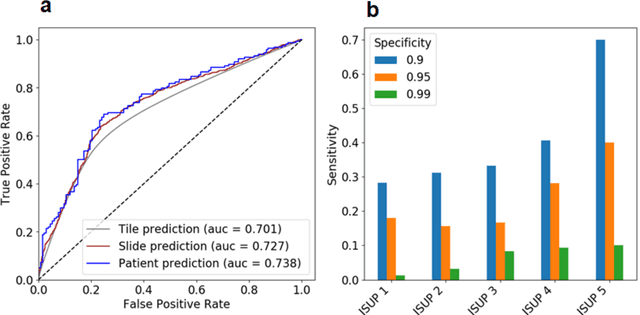

Background: Transrectal ultrasound guided systematic biopsies of the prostate is a routine procedure to establish a prostate cancer diagnosis. However, the 10-12 prostate core biopsies only sample a relatively small volume of the prostate, and tumour lesions in regions between biopsy cores can be missed, leading to a well-known low sensitivity to detect clinically relevant cancer. As a proof-of-principle, we developed and validated a deep convolutional neural network model to distinguish between morphological patterns in benign prostate biopsy whole slide images from men with and without established cancer. Methods: This study included 14,354 hematoxylin and eosin stained whole slide images from benign prostate biopsies from 1,508 men in two groups: men without an established prostate cancer (PCa) diagnosis and men with at least one core biopsy diagnosed with PCa. 80% of the participants were assigned as training data and used for model optimization (1,211 men), and the remaining 20% (297 men) as a held-out test set used to evaluate model performance. An ensemble of 10 deep convolutional neural network models was optimized for classification of biopsies from men with and without established cancer. Hyperparameter optimization and model selection was performed by cross-validation in the training data . Results: Area under the receiver operating characteristic curve (ROC-AUC) was estimated as 0.727 (bootstrap 95% CI: 0.708-0.745) on biopsy level and 0.738 (bootstrap 95% CI: 0.682 - 0.796) on man level. At a specificity of 0.9 the model had an estimated sensitivity of 0.348. Conclusion: The developed model has the ability to detect men with risk of missed PCa due to under-sampling of the prostate. The proposed model has the potential to reduce the number of false negative cases in routine systematic prostate biopsies and to indicate men who could benefit from MRI-guided re-biopsy.