 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTranscriptome-supervised classification of tissue morphology using deep learning
Dec 07, 2023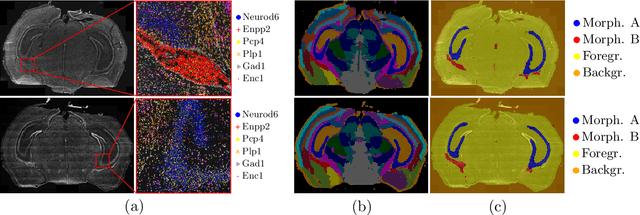

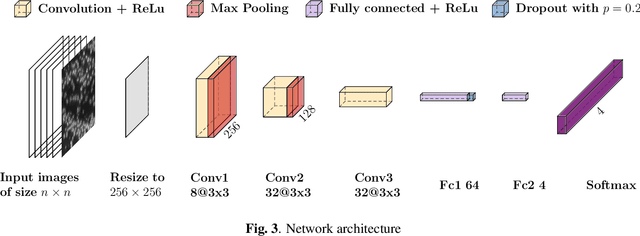
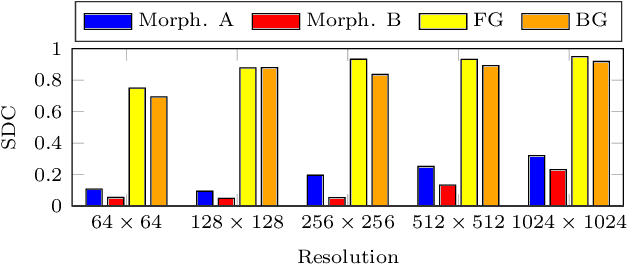
Deep learning has proven to successfully learn variations in tissue and cell morphology. Training of such models typically relies on expensive manual annotations. Here we conjecture that spatially resolved gene expression, e.i., the transcriptome, can be used as an alternative to manual annotations. In particular, we trained five convolutional neural networks with patches of different size extracted from locations defined by spatially resolved gene expression. The network is trained to classify tissue morphology related to two different genes, general tissue, as well as background, on an image of fluorescence stained nuclei in a mouse brain coronal section. Performance is evaluated on an independent tissue section from a different mouse brain, reaching an average Dice score of 0.51. Results may indicate that novel techniques for spatially resolved transcriptomics together with deep learning may provide a unique and unbiased way to find genotype-phenotype relationships.
* Accepted for publication at IEEE International Symposium on Biomedical Imaging (ISBI) 2020
The ACROBAT 2022 Challenge: Automatic Registration Of Breast Cancer Tissue
May 29, 2023The alignment of tissue between histopathological whole-slide-images (WSI) is crucial for research and clinical applications. Advances in computing, deep learning, and availability of large WSI datasets have revolutionised WSI analysis. Therefore, the current state-of-the-art in WSI registration is unclear. To address this, we conducted the ACROBAT challenge, based on the largest WSI registration dataset to date, including 4,212 WSIs from 1,152 breast cancer patients. The challenge objective was to align WSIs of tissue that was stained with routine diagnostic immunohistochemistry to its H&E-stained counterpart. We compare the performance of eight WSI registration algorithms, including an investigation of the impact of different WSI properties and clinical covariates. We find that conceptually distinct WSI registration methods can lead to highly accurate registration performances and identify covariates that impact performances across methods. These results establish the current state-of-the-art in WSI registration and guide researchers in selecting and developing methods.
ACROBAT -- a multi-stain breast cancer histological whole-slide-image data set from routine diagnostics for computational pathology
Nov 24, 2022The analysis of FFPE tissue sections stained with haematoxylin and eosin (H&E) or immunohistochemistry (IHC) is an essential part of the pathologic assessment of surgically resected breast cancer specimens. IHC staining has been broadly adopted into diagnostic guidelines and routine workflows to manually assess status and scoring of several established biomarkers, including ER, PGR, HER2 and KI67. However, this is a task that can also be facilitated by computational pathology image analysis methods. The research in computational pathology has recently made numerous substantial advances, often based on publicly available whole slide image (WSI) data sets. However, the field is still considerably limited by the sparsity of public data sets. In particular, there are no large, high quality publicly available data sets with WSIs of matching IHC and H&E-stained tissue sections. Here, we publish the currently largest publicly available data set of WSIs of tissue sections from surgical resection specimens from female primary breast cancer patients with matched WSIs of corresponding H&E and IHC-stained tissue, consisting of 4,212 WSIs from 1,153 patients. The primary purpose of the data set was to facilitate the ACROBAT WSI registration challenge, aiming at accurately aligning H&E and IHC images. For research in the area of image registration, automatic quantitative feedback on registration algorithm performance remains available through the ACROBAT challenge website, based on more than 37,000 manually annotated landmark pairs from 13 annotators. Beyond registration, this data set has the potential to enable many different avenues of computational pathology research, including stain-guided learning, virtual staining, unsupervised pre-training, artefact detection and stain-independent models.
Pathologist-Level Grading of Prostate Biopsies with Artificial Intelligence
Jul 02, 2019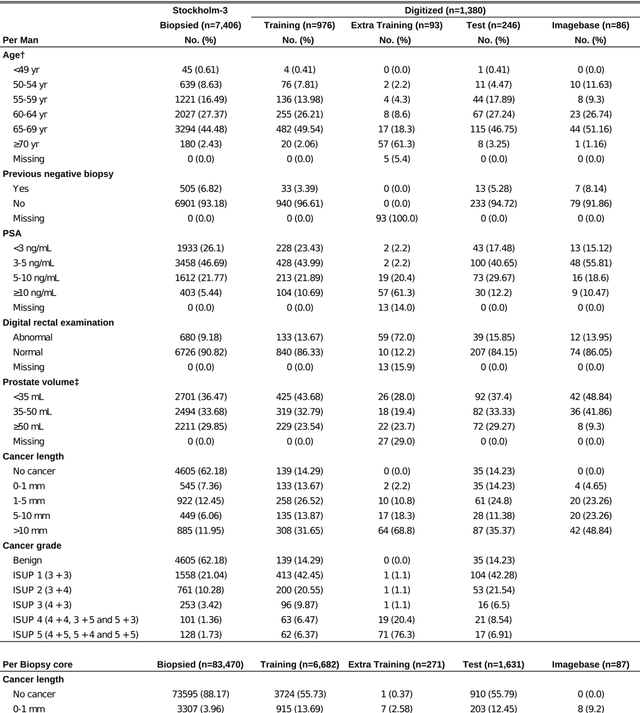
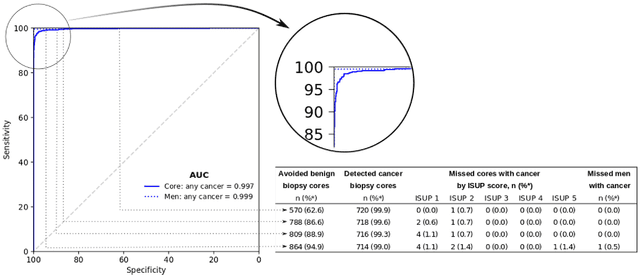
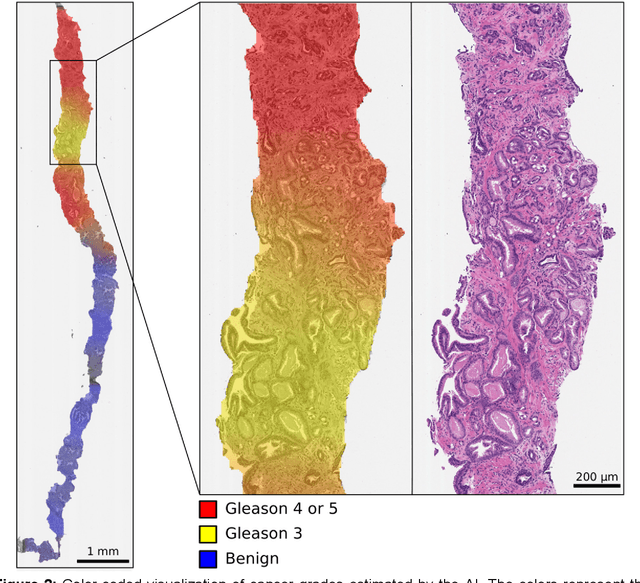
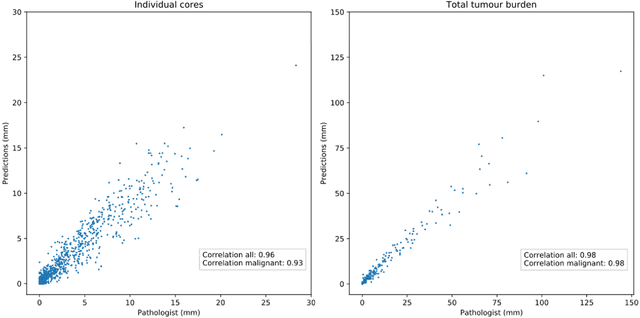
Background: An increasing volume of prostate biopsies and a world-wide shortage of uro-pathologists puts a strain on pathology departments. Additionally, the high intra- and inter-observer variability in grading can result in over- and undertreatment of prostate cancer. Artificial intelligence (AI) methods may alleviate these problems by assisting pathologists to reduce workload and harmonize grading. Methods: We digitized 6,682 needle biopsies from 976 participants in the population based STHLM3 diagnostic study to train deep neural networks for assessing prostate biopsies. The networks were evaluated by predicting the presence, extent, and Gleason grade of malignant tissue for an independent test set comprising 1,631 biopsies from 245 men. We additionally evaluated grading performance on 87 biopsies individually graded by 23 experienced urological pathologists from the International Society of Urological Pathology. We assessed discriminatory performance by receiver operating characteristics (ROC) and tumor extent predictions by correlating predicted millimeter cancer length against measurements by the reporting pathologist. We quantified the concordance between grades assigned by the AI and the expert urological pathologists using Cohen's kappa. Results: The performance of the AI to detect and grade cancer in prostate needle biopsy samples was comparable to that of international experts in prostate pathology. The AI achieved an area under the ROC curve of 0.997 for distinguishing between benign and malignant biopsy cores, and 0.999 for distinguishing between men with or without prostate cancer. The correlation between millimeter cancer predicted by the AI and assigned by the reporting pathologist was 0.96. For assigning Gleason grades, the AI achieved an average pairwise kappa of 0.62. This was within the range of the corresponding values for the expert pathologists (0.60 to 0.73).
Whole slide image registration for the study of tumor heterogeneity
Jan 24, 2019



Consecutive thin sections of tissue samples make it possible to study local variation in e.g. protein expression and tumor heterogeneity by staining for a new protein in each section. In order to compare and correlate patterns of different proteins, the images have to be registered with high accuracy. The problem we want to solve is registration of gigapixel whole slide images (WSI). This presents 3 challenges: (i) Images are very large; (ii) Thin sections result in artifacts that make global affine registration prone to very large local errors; (iii) Local affine registration is required to preserve correct tissue morphology (local size, shape and texture). In our approach we compare WSI registration based on automatic and manual feature selection on either the full image or natural sub-regions (as opposed to square tiles). Working with natural sub-regions, in an interactive tool makes it possible to exclude regions containing scientifically irrelevant information. We also present a new way to visualize local registration quality by a Registration Confidence Map (RCM). With this method, intra-tumor heterogeneity and charateristics of the tumor microenvironment can be observed and quantified.
* MICCAI2018 - Computational Pathology and Ophthalmic Medical Image Analysis - COMPAY