 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTranscriptome-wide prediction of prostate cancer gene expression from histopathology images using co-expression based convolutional neural networks
Paper and Code
Apr 19, 2021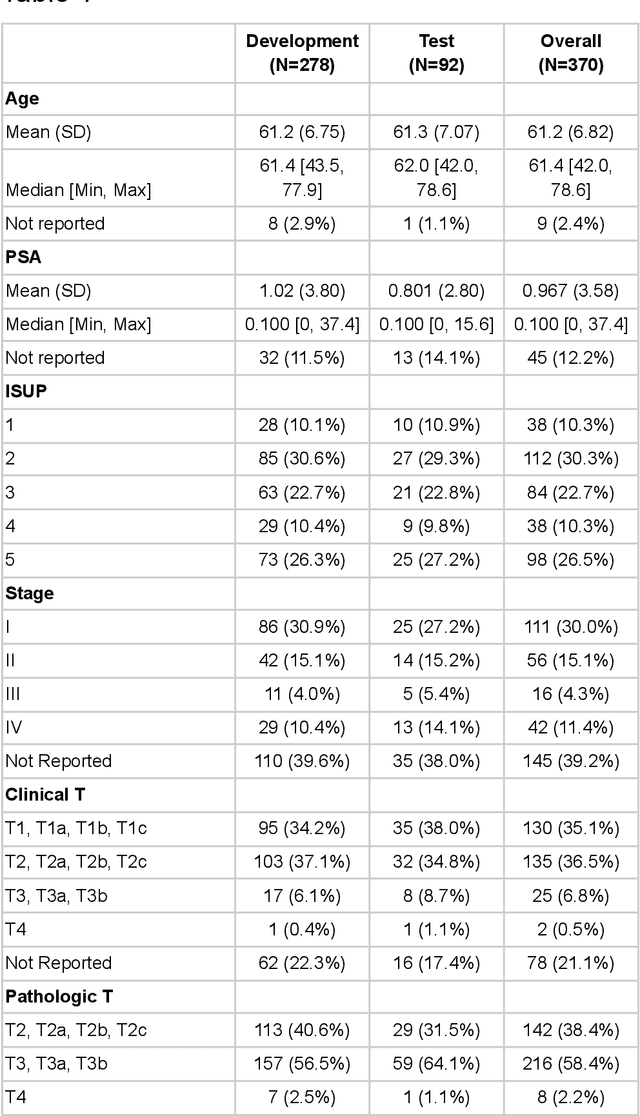
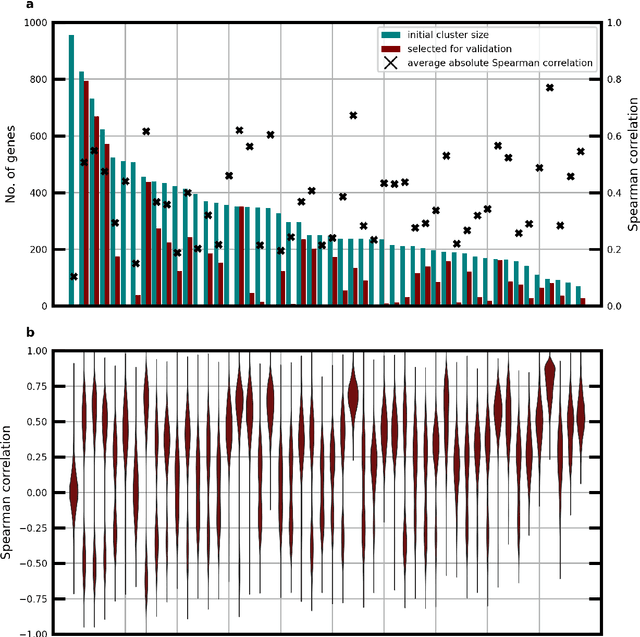
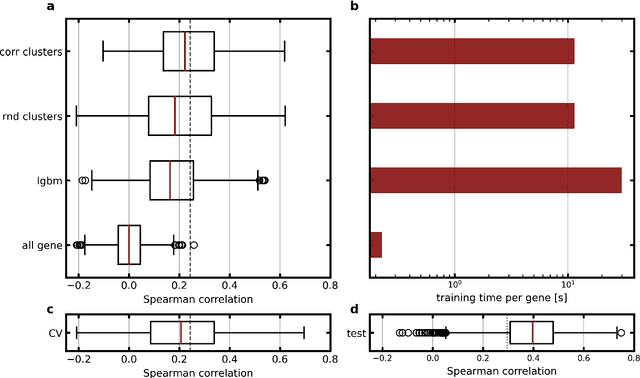
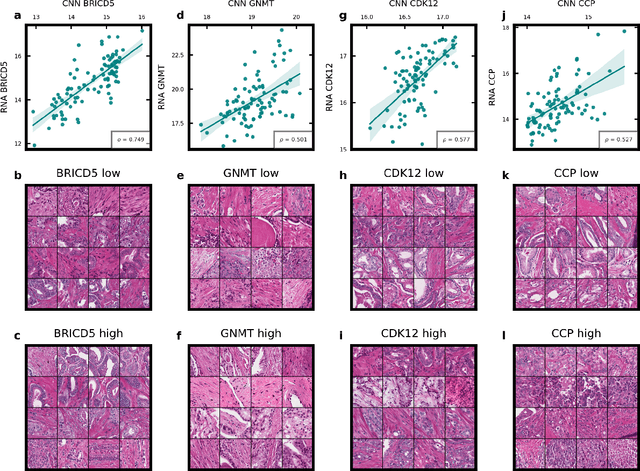
Molecular phenotyping by gene expression profiling is common in contemporary cancer research and in molecular diagnostics. However, molecular profiling remains costly and resource intense to implement, and is just starting to be introduced into clinical diagnostics. Molecular changes, including genetic alterations and gene expression changes, occuring in tumors cause morphological changes in tissue, which can be observed on the microscopic level. The relationship between morphological patterns and some of the molecular phenotypes can be exploited to predict molecular phenotypes directly from routine haematoxylin and eosin (H&E) stained whole slide images (WSIs) using deep convolutional neural networks (CNNs). In this study, we propose a new, computationally efficient approach for disease specific modelling of relationships between morphology and gene expression, and we conducted the first transcriptome-wide analysis in prostate cancer, using CNNs to predict bulk RNA-sequencing estimates from WSIs of H&E stained tissue. The work is based on the TCGA PRAD study and includes both WSIs and RNA-seq data for 370 patients. Out of 15586 protein coding and sufficiently frequently expressed transcripts, 6618 had predicted expression significantly associated with RNA-seq estimates (FDR-adjusted p-value < 1*10-4) in a cross-validation. 5419 (81.9%) of these were subsequently validated in a held-out test set. We also demonstrate the ability to predict a prostate cancer specific cell cycle progression score directly from WSIs. These findings suggest that contemporary computer vision models offer an inexpensive and scalable solution for prediction of gene expression phenotypes directly from WSIs, providing opportunity for cost-effective large-scale research studies and molecular diagnostics.