 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOne-Shot Affordance Grounding of Deformable Objects in Egocentric Organizing Scenes
Mar 03, 2025Deformable object manipulation in robotics presents significant challenges due to uncertainties in component properties, diverse configurations, visual interference, and ambiguous prompts. These factors complicate both perception and control tasks. To address these challenges, we propose a novel method for One-Shot Affordance Grounding of Deformable Objects (OS-AGDO) in egocentric organizing scenes, enabling robots to recognize previously unseen deformable objects with varying colors and shapes using minimal samples. Specifically, we first introduce the Deformable Object Semantic Enhancement Module (DefoSEM), which enhances hierarchical understanding of the internal structure and improves the ability to accurately identify local features, even under conditions of weak component information. Next, we propose the ORB-Enhanced Keypoint Fusion Module (OEKFM), which optimizes feature extraction of key components by leveraging geometric constraints and improves adaptability to diversity and visual interference. Additionally, we propose an instance-conditional prompt based on image data and task context, effectively mitigates the issue of region ambiguity caused by prompt words. To validate these methods, we construct a diverse real-world dataset, AGDDO15, which includes 15 common types of deformable objects and their associated organizational actions. Experimental results demonstrate that our approach significantly outperforms state-of-the-art methods, achieving improvements of 6.2%, 3.2%, and 2.9% in KLD, SIM, and NSS metrics, respectively, while exhibiting high generalization performance. Source code and benchmark dataset will be publicly available at https://github.com/Dikay1/OS-AGDO.
Physical Color Calibration of Digital Pathology Scanners for Robust Artificial Intelligence Assisted Cancer Diagnosis
Jul 07, 2023



The potential of artificial intelligence (AI) in digital pathology is limited by technical inconsistencies in the production of whole slide images (WSIs), leading to degraded AI performance and posing a challenge for widespread clinical application as fine-tuning algorithms for each new site is impractical. Changes in the imaging workflow can also lead to compromised diagnoses and patient safety risks. We evaluated whether physical color calibration of scanners can standardize WSI appearance and enable robust AI performance. We employed a color calibration slide in four different laboratories and evaluated its impact on the performance of an AI system for prostate cancer diagnosis on 1,161 WSIs. Color standardization resulted in consistently improved AI model calibration and significant improvements in Gleason grading performance. The study demonstrates that physical color calibration provides a potential solution to the variation introduced by different scanners, making AI-based cancer diagnostics more reliable and applicable in clinical settings.
Using deep learning to detect patients at risk for prostate cancer despite benign biopsies
Jul 31, 2021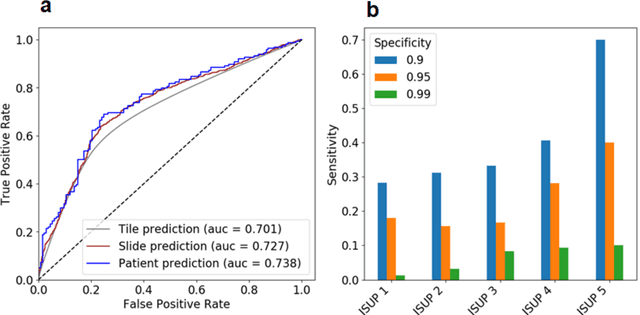

Background: Transrectal ultrasound guided systematic biopsies of the prostate is a routine procedure to establish a prostate cancer diagnosis. However, the 10-12 prostate core biopsies only sample a relatively small volume of the prostate, and tumour lesions in regions between biopsy cores can be missed, leading to a well-known low sensitivity to detect clinically relevant cancer. As a proof-of-principle, we developed and validated a deep convolutional neural network model to distinguish between morphological patterns in benign prostate biopsy whole slide images from men with and without established cancer. Methods: This study included 14,354 hematoxylin and eosin stained whole slide images from benign prostate biopsies from 1,508 men in two groups: men without an established prostate cancer (PCa) diagnosis and men with at least one core biopsy diagnosed with PCa. 80% of the participants were assigned as training data and used for model optimization (1,211 men), and the remaining 20% (297 men) as a held-out test set used to evaluate model performance. An ensemble of 10 deep convolutional neural network models was optimized for classification of biopsies from men with and without established cancer. Hyperparameter optimization and model selection was performed by cross-validation in the training data . Results: Area under the receiver operating characteristic curve (ROC-AUC) was estimated as 0.727 (bootstrap 95% CI: 0.708-0.745) on biopsy level and 0.738 (bootstrap 95% CI: 0.682 - 0.796) on man level. At a specificity of 0.9 the model had an estimated sensitivity of 0.348. Conclusion: The developed model has the ability to detect men with risk of missed PCa due to under-sampling of the prostate. The proposed model has the potential to reduce the number of false negative cases in routine systematic prostate biopsies and to indicate men who could benefit from MRI-guided re-biopsy.
Transcriptome-wide prediction of prostate cancer gene expression from histopathology images using co-expression based convolutional neural networks
Apr 19, 2021
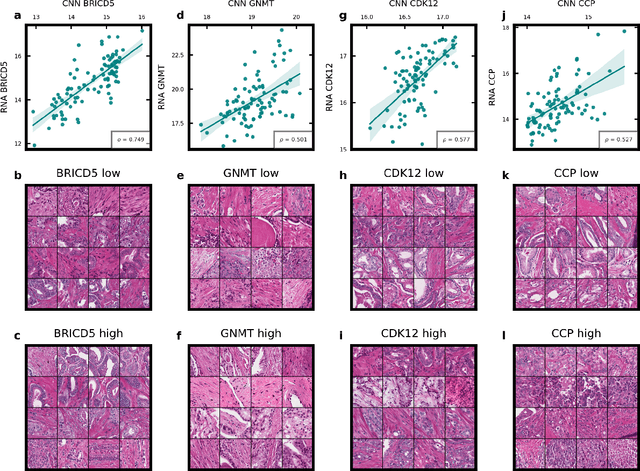
Molecular phenotyping by gene expression profiling is common in contemporary cancer research and in molecular diagnostics. However, molecular profiling remains costly and resource intense to implement, and is just starting to be introduced into clinical diagnostics. Molecular changes, including genetic alterations and gene expression changes, occuring in tumors cause morphological changes in tissue, which can be observed on the microscopic level. The relationship between morphological patterns and some of the molecular phenotypes can be exploited to predict molecular phenotypes directly from routine haematoxylin and eosin (H&E) stained whole slide images (WSIs) using deep convolutional neural networks (CNNs). In this study, we propose a new, computationally efficient approach for disease specific modelling of relationships between morphology and gene expression, and we conducted the first transcriptome-wide analysis in prostate cancer, using CNNs to predict bulk RNA-sequencing estimates from WSIs of H&E stained tissue. The work is based on the TCGA PRAD study and includes both WSIs and RNA-seq data for 370 patients. Out of 15586 protein coding and sufficiently frequently expressed transcripts, 6618 had predicted expression significantly associated with RNA-seq estimates (FDR-adjusted p-value < 1*10-4) in a cross-validation. 5419 (81.9%) of these were subsequently validated in a held-out test set. We also demonstrate the ability to predict a prostate cancer specific cell cycle progression score directly from WSIs. These findings suggest that contemporary computer vision models offer an inexpensive and scalable solution for prediction of gene expression phenotypes directly from WSIs, providing opportunity for cost-effective large-scale research studies and molecular diagnostics.
Predicting molecular phenotypes from histopathology images: a transcriptome-wide expression-morphology analysis in breast cancer
Sep 18, 2020
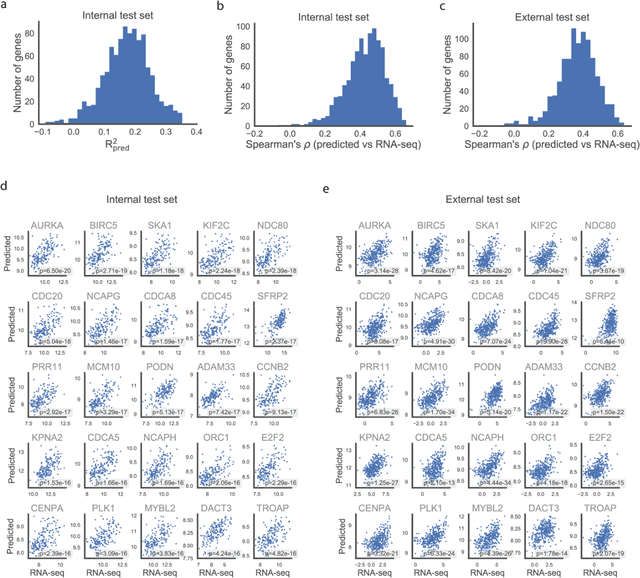
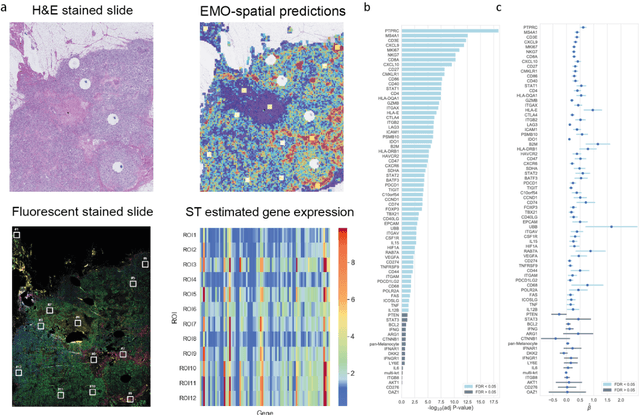
Molecular phenotyping is central in cancer precision medicine, but remains costly and standard methods only provide a tumour average profile. Microscopic morphological patterns observable in histopathology sections from tumours are determined by the underlying molecular phenotype and associated with clinical factors. The relationship between morphology and molecular phenotype has a potential to be exploited for prediction of the molecular phenotype from the morphology visible in histopathology images. We report the first transcriptome-wide Expression-MOrphology (EMO) analysis in breast cancer, where gene-specific models were optimised and validated for prediction of mRNA expression both as a tumour average and in spatially resolved manner. Individual deep convolutional neural networks (CNNs) were optimised to predict the expression of 17,695 genes from hematoxylin and eosin (HE) stained whole slide images (WSIs). Predictions for 9,334 (52.75%) genes were significantly associated with RNA-sequencing estimates (FDR adjusted p-value < 0.05). 1,011 of the genes were brought forward for validation, with 876 (87%) and 908 (90%) successfully replicated in internal and external test data, respectively. Predicted spatial intra-tumour variabilities in expression were validated in 76 genes, out of which 59 (77.6%) had a significant association (FDR adjusted p-value < 0.05) with spatial transcriptomics estimates. These results suggest that the proposed methodology can be applied to predict both tumour average gene expression and intra-tumour spatial expression directly from morphology, thus providing a scalable approach to characterise intra-tumour heterogeneity.