 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe ACROBAT 2022 Challenge: Automatic Registration Of Breast Cancer Tissue
May 29, 2023The alignment of tissue between histopathological whole-slide-images (WSI) is crucial for research and clinical applications. Advances in computing, deep learning, and availability of large WSI datasets have revolutionised WSI analysis. Therefore, the current state-of-the-art in WSI registration is unclear. To address this, we conducted the ACROBAT challenge, based on the largest WSI registration dataset to date, including 4,212 WSIs from 1,152 breast cancer patients. The challenge objective was to align WSIs of tissue that was stained with routine diagnostic immunohistochemistry to its H&E-stained counterpart. We compare the performance of eight WSI registration algorithms, including an investigation of the impact of different WSI properties and clinical covariates. We find that conceptually distinct WSI registration methods can lead to highly accurate registration performances and identify covariates that impact performances across methods. These results establish the current state-of-the-art in WSI registration and guide researchers in selecting and developing methods.
Increasing the usefulness of already existing annotations through WSI registration
Mar 12, 2023Computational pathology methods have the potential to improve access to precision medicine, as well as the reproducibility and accuracy of pathological diagnoses. Particularly the analysis of whole-slide-images (WSIs) of immunohistochemically (IHC) stained tissue sections could benefit from computational pathology methods. However, scoring biomarkers such as KI67 in IHC WSIs often necessitates the detection of areas of invasive cancer. Training cancer detection models often requires annotations, which is time-consuming and therefore costly. Currently, cancer regions are typically annotated in WSIs of haematoxylin and eosin (H&E) stained tissue sections. In this study, we investigate the possibility to register annotations that were made in H&E WSIs to their IHC counterparts. Two pathologists annotated regions of invasive cancer in WSIs of 272 breast cancer cases. For each case, a matched H&E and KI67 WSI are available, resulting in 544 WSIs with invasive cancer annotations. We find that cancer detection CNNs that were trained with annotations registered from the H&E to the KI67 WSIs only differ slightly in calibration but not in performance compared to cancer detection models trained on annotations made directly in the KI67 WSIs in a test set consisting of 54 cases. The mean slide-level AUROC is 0.974 [0.964, 0.982] for models trained with the KI67 annotations and 0.974 [0.965, 0.982] for models trained using registered annotations. This indicates that WSI registration has the potential to reduce the need for IHC-specific annotations. This could significantly increase the usefulness of already existing annotations.
ACROBAT -- a multi-stain breast cancer histological whole-slide-image data set from routine diagnostics for computational pathology
Nov 24, 2022The analysis of FFPE tissue sections stained with haematoxylin and eosin (H&E) or immunohistochemistry (IHC) is an essential part of the pathologic assessment of surgically resected breast cancer specimens. IHC staining has been broadly adopted into diagnostic guidelines and routine workflows to manually assess status and scoring of several established biomarkers, including ER, PGR, HER2 and KI67. However, this is a task that can also be facilitated by computational pathology image analysis methods. The research in computational pathology has recently made numerous substantial advances, often based on publicly available whole slide image (WSI) data sets. However, the field is still considerably limited by the sparsity of public data sets. In particular, there are no large, high quality publicly available data sets with WSIs of matching IHC and H&E-stained tissue sections. Here, we publish the currently largest publicly available data set of WSIs of tissue sections from surgical resection specimens from female primary breast cancer patients with matched WSIs of corresponding H&E and IHC-stained tissue, consisting of 4,212 WSIs from 1,153 patients. The primary purpose of the data set was to facilitate the ACROBAT WSI registration challenge, aiming at accurately aligning H&E and IHC images. For research in the area of image registration, automatic quantitative feedback on registration algorithm performance remains available through the ACROBAT challenge website, based on more than 37,000 manually annotated landmark pairs from 13 annotators. Beyond registration, this data set has the potential to enable many different avenues of computational pathology research, including stain-guided learning, virtual staining, unsupervised pre-training, artefact detection and stain-independent models.
Using deep learning to detect patients at risk for prostate cancer despite benign biopsies
Jul 31, 2021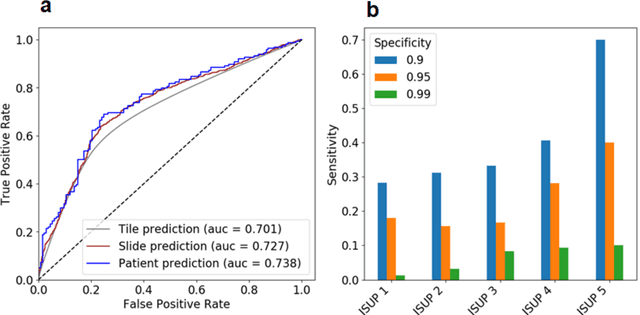

Background: Transrectal ultrasound guided systematic biopsies of the prostate is a routine procedure to establish a prostate cancer diagnosis. However, the 10-12 prostate core biopsies only sample a relatively small volume of the prostate, and tumour lesions in regions between biopsy cores can be missed, leading to a well-known low sensitivity to detect clinically relevant cancer. As a proof-of-principle, we developed and validated a deep convolutional neural network model to distinguish between morphological patterns in benign prostate biopsy whole slide images from men with and without established cancer. Methods: This study included 14,354 hematoxylin and eosin stained whole slide images from benign prostate biopsies from 1,508 men in two groups: men without an established prostate cancer (PCa) diagnosis and men with at least one core biopsy diagnosed with PCa. 80% of the participants were assigned as training data and used for model optimization (1,211 men), and the remaining 20% (297 men) as a held-out test set used to evaluate model performance. An ensemble of 10 deep convolutional neural network models was optimized for classification of biopsies from men with and without established cancer. Hyperparameter optimization and model selection was performed by cross-validation in the training data . Results: Area under the receiver operating characteristic curve (ROC-AUC) was estimated as 0.727 (bootstrap 95% CI: 0.708-0.745) on biopsy level and 0.738 (bootstrap 95% CI: 0.682 - 0.796) on man level. At a specificity of 0.9 the model had an estimated sensitivity of 0.348. Conclusion: The developed model has the ability to detect men with risk of missed PCa due to under-sampling of the prostate. The proposed model has the potential to reduce the number of false negative cases in routine systematic prostate biopsies and to indicate men who could benefit from MRI-guided re-biopsy.
Predicting molecular phenotypes from histopathology images: a transcriptome-wide expression-morphology analysis in breast cancer
Sep 18, 2020
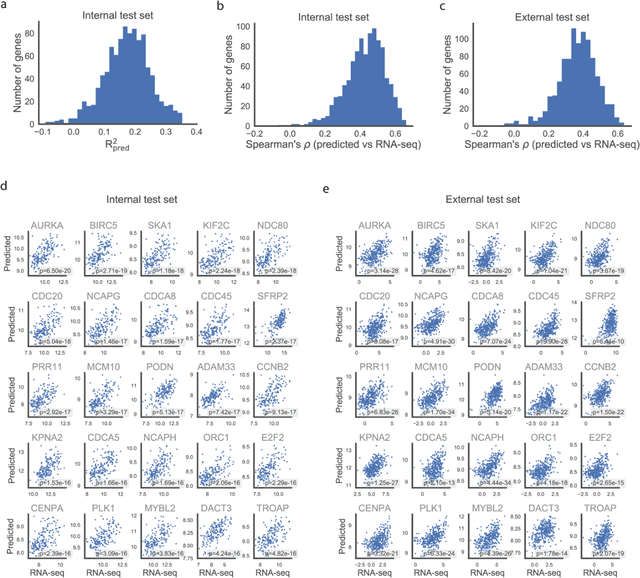
Molecular phenotyping is central in cancer precision medicine, but remains costly and standard methods only provide a tumour average profile. Microscopic morphological patterns observable in histopathology sections from tumours are determined by the underlying molecular phenotype and associated with clinical factors. The relationship between morphology and molecular phenotype has a potential to be exploited for prediction of the molecular phenotype from the morphology visible in histopathology images. We report the first transcriptome-wide Expression-MOrphology (EMO) analysis in breast cancer, where gene-specific models were optimised and validated for prediction of mRNA expression both as a tumour average and in spatially resolved manner. Individual deep convolutional neural networks (CNNs) were optimised to predict the expression of 17,695 genes from hematoxylin and eosin (HE) stained whole slide images (WSIs). Predictions for 9,334 (52.75%) genes were significantly associated with RNA-sequencing estimates (FDR adjusted p-value < 0.05). 1,011 of the genes were brought forward for validation, with 876 (87%) and 908 (90%) successfully replicated in internal and external test data, respectively. Predicted spatial intra-tumour variabilities in expression were validated in 76 genes, out of which 59 (77.6%) had a significant association (FDR adjusted p-value < 0.05) with spatial transcriptomics estimates. These results suggest that the proposed methodology can be applied to predict both tumour average gene expression and intra-tumour spatial expression directly from morphology, thus providing a scalable approach to characterise intra-tumour heterogeneity.