 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePredicting molecular phenotypes from histopathology images: a transcriptome-wide expression-morphology analysis in breast cancer
Sep 18, 2020
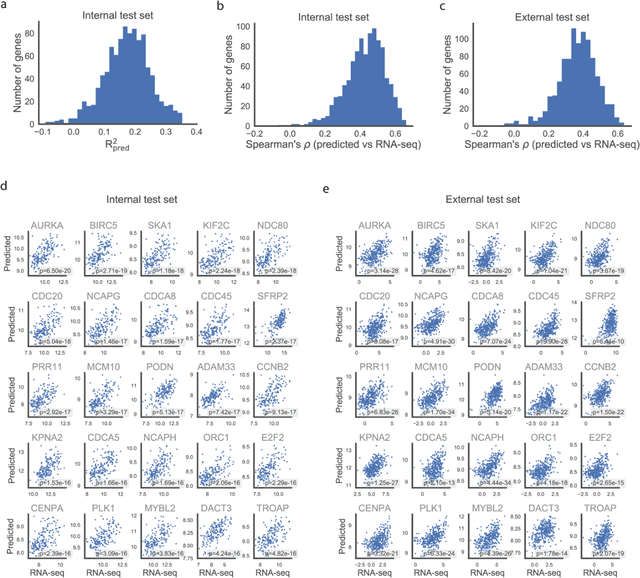
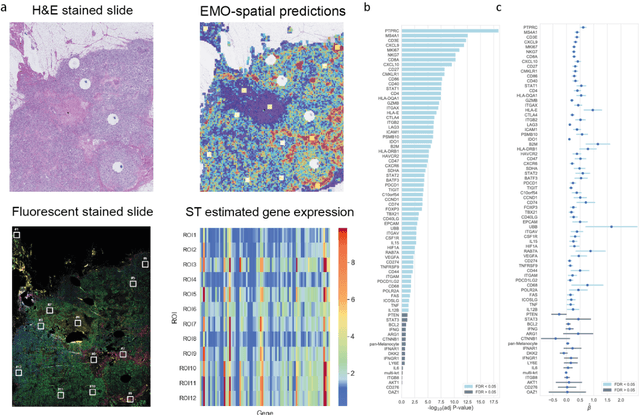
Molecular phenotyping is central in cancer precision medicine, but remains costly and standard methods only provide a tumour average profile. Microscopic morphological patterns observable in histopathology sections from tumours are determined by the underlying molecular phenotype and associated with clinical factors. The relationship between morphology and molecular phenotype has a potential to be exploited for prediction of the molecular phenotype from the morphology visible in histopathology images. We report the first transcriptome-wide Expression-MOrphology (EMO) analysis in breast cancer, where gene-specific models were optimised and validated for prediction of mRNA expression both as a tumour average and in spatially resolved manner. Individual deep convolutional neural networks (CNNs) were optimised to predict the expression of 17,695 genes from hematoxylin and eosin (HE) stained whole slide images (WSIs). Predictions for 9,334 (52.75%) genes were significantly associated with RNA-sequencing estimates (FDR adjusted p-value < 0.05). 1,011 of the genes were brought forward for validation, with 876 (87%) and 908 (90%) successfully replicated in internal and external test data, respectively. Predicted spatial intra-tumour variabilities in expression were validated in 76 genes, out of which 59 (77.6%) had a significant association (FDR adjusted p-value < 0.05) with spatial transcriptomics estimates. These results suggest that the proposed methodology can be applied to predict both tumour average gene expression and intra-tumour spatial expression directly from morphology, thus providing a scalable approach to characterise intra-tumour heterogeneity.