 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCoFrGeNet: Continued Fraction Architectures for Language Generation
Jan 29, 2026Transformers are arguably the preferred architecture for language generation. In this paper, inspired by continued fractions, we introduce a new function class for generative modeling. The architecture family implementing this function class is named CoFrGeNets - Continued Fraction Generative Networks. We design novel architectural components based on this function class that can replace Multi-head Attention and Feed-Forward Networks in Transformer blocks while requiring much fewer parameters. We derive custom gradient formulations to optimize the proposed components more accurately and efficiently than using standard PyTorch-based gradients. Our components are a plug-in replacement requiring little change in training or inference procedures that have already been put in place for Transformer-based models thus making our approach easy to incorporate in large industrial workflows. We experiment on two very different transformer architectures GPT2-xl (1.5B) and Llama3 (3.2B), where the former we pre-train on OpenWebText and GneissWeb, while the latter we pre-train on the docling data mix which consists of nine different datasets. Results show that the performance on downstream classification, Q\& A, reasoning and text understanding tasks of our models is competitive and sometimes even superior to the original models with $\frac{2}{3}$ to $\frac{1}{2}$ the parameters and shorter pre-training time. We believe that future implementations customized to hardware will further bring out the true potential of our architectures.
GP-MoLFormer-Sim: Test Time Molecular Optimization through Contextual Similarity Guidance
Jun 05, 2025The ability to design molecules while preserving similarity to a target molecule and/or property is crucial for various applications in drug discovery, chemical design, and biology. We introduce in this paper an efficient training-free method for navigating and sampling from the molecular space with a generative Chemical Language Model (CLM), while using the molecular similarity to the target as a guide. Our method leverages the contextual representations learned from the CLM itself to estimate the molecular similarity, which is then used to adjust the autoregressive sampling strategy of the CLM. At each step of the decoding process, the method tracks the distance of the current generations from the target and updates the logits to encourage the preservation of similarity in generations. We implement the method using a recently proposed $\sim$47M parameter SMILES-based CLM, GP-MoLFormer, and therefore refer to the method as GP-MoLFormer-Sim, which enables a test-time update of the deep generative policy to reflect the contextual similarity to a set of guide molecules. The method is further integrated into a genetic algorithm (GA) and tested on a set of standard molecular optimization benchmarks involving property optimization, molecular rediscovery, and structure-based drug design. Results show that, GP-MoLFormer-Sim, combined with GA (GP-MoLFormer-Sim+GA) outperforms existing training-free baseline methods, when the oracle remains black-box. The findings in this work are a step forward in understanding and guiding the generative mechanisms of CLMs.
Aligning Protein Conformation Ensemble Generation with Physical Feedback
May 30, 2025Protein dynamics play a crucial role in protein biological functions and properties, and their traditional study typically relies on time-consuming molecular dynamics (MD) simulations conducted in silico. Recent advances in generative modeling, particularly denoising diffusion models, have enabled efficient accurate protein structure prediction and conformation sampling by learning distributions over crystallographic structures. However, effectively integrating physical supervision into these data-driven approaches remains challenging, as standard energy-based objectives often lead to intractable optimization. In this paper, we introduce Energy-based Alignment (EBA), a method that aligns generative models with feedback from physical models, efficiently calibrating them to appropriately balance conformational states based on their energy differences. Experimental results on the MD ensemble benchmark demonstrate that EBA achieves state-of-the-art performance in generating high-quality protein ensembles. By improving the physical plausibility of generated structures, our approach enhances model predictions and holds promise for applications in structural biology and drug discovery.
Larimar: Large Language Models with Episodic Memory Control
Mar 18, 2024Efficient and accurate updating of knowledge stored in Large Language Models (LLMs) is one of the most pressing research challenges today. This paper presents Larimar - a novel, brain-inspired architecture for enhancing LLMs with a distributed episodic memory. Larimar's memory allows for dynamic, one-shot updates of knowledge without the need for computationally expensive re-training or fine-tuning. Experimental results on multiple fact editing benchmarks demonstrate that Larimar attains accuracy comparable to most competitive baselines, even in the challenging sequential editing setup, but also excels in speed - yielding speed-ups of 4-10x depending on the base LLM - as well as flexibility due to the proposed architecture being simple, LLM-agnostic, and hence general. We further provide mechanisms for selective fact forgetting and input context length generalization with Larimar and show their effectiveness.
ProtIR: Iterative Refinement between Retrievers and Predictors for Protein Function Annotation
Feb 10, 2024
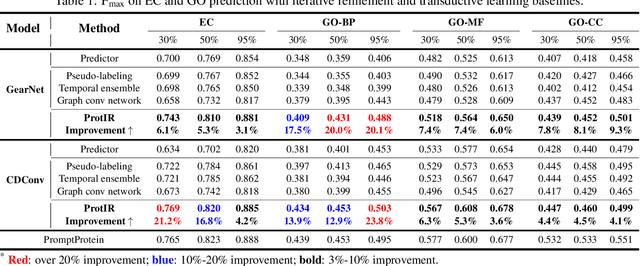
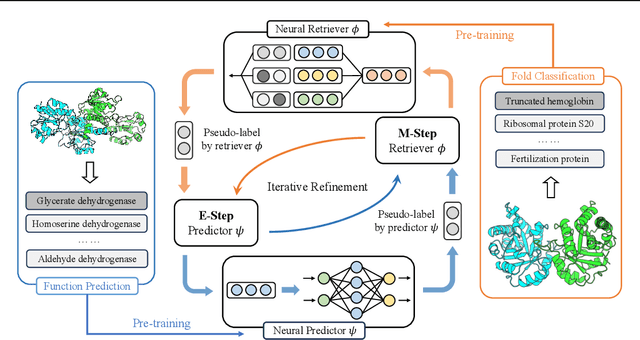

Protein function annotation is an important yet challenging task in biology. Recent deep learning advancements show significant potential for accurate function prediction by learning from protein sequences and structures. Nevertheless, these predictor-based methods often overlook the modeling of protein similarity, an idea commonly employed in traditional approaches using sequence or structure retrieval tools. To fill this gap, we first study the effect of inter-protein similarity modeling by benchmarking retriever-based methods against predictors on protein function annotation tasks. Our results show that retrievers can match or outperform predictors without large-scale pre-training. Building on these insights, we introduce a novel variational pseudo-likelihood framework, ProtIR, designed to improve function predictors by incorporating inter-protein similarity modeling. This framework iteratively refines knowledge between a function predictor and retriever, thereby combining the strengths of both predictors and retrievers. ProtIR showcases around 10% improvement over vanilla predictor-based methods. Besides, it achieves performance on par with protein language model-based methods, yet without the need for massive pre-training, highlighting the efficacy of our framework. Code will be released upon acceptance.
Structure-Informed Protein Language Model
Feb 07, 2024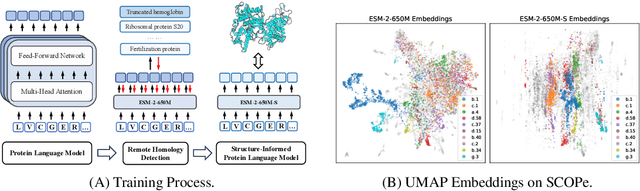

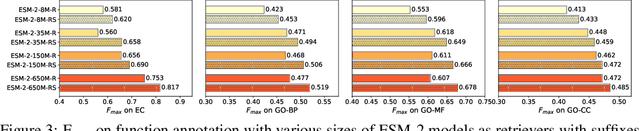
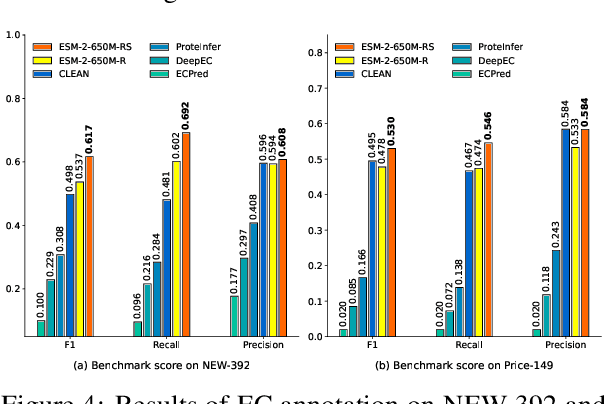
Protein language models are a powerful tool for learning protein representations through pre-training on vast protein sequence datasets. However, traditional protein language models lack explicit structural supervision, despite its relevance to protein function. To address this issue, we introduce the integration of remote homology detection to distill structural information into protein language models without requiring explicit protein structures as input. We evaluate the impact of this structure-informed training on downstream protein function prediction tasks. Experimental results reveal consistent improvements in function annotation accuracy for EC number and GO term prediction. Performance on mutant datasets, however, varies based on the relationship between targeted properties and protein structures. This underscores the importance of considering this relationship when applying structure-aware training to protein function prediction tasks. Code and model weights are available at https://github.com/DeepGraphLearning/esm-s.
Equivariant Few-Shot Learning from Pretrained Models
May 17, 2023Efficient transfer learning algorithms are key to the success of foundation models on diverse downstream tasks even with limited data. Recent works of \cite{basu2022equi} and \cite{kaba2022equivariance} propose group averaging (\textit{equitune}) and optimization-based methods, respectively, over features from group-transformed inputs to obtain equivariant outputs from non-equivariant neural networks. While \cite{kaba2022equivariance} are only concerned with training from scratch, we find that equitune performs poorly on equivariant zero-shot tasks despite good finetuning results. We hypothesize that this is because pretrained models provide better quality features for certain transformations than others and simply averaging them is deleterious. Hence, we propose $\lambda$-\textit{equitune} that averages the features using \textit{importance weights}, $\lambda$s. These weights are learned directly from the data using a small neural network, leading to excellent zero-shot and finetuned results that outperform equitune. Further, we prove that $\lambda$-equitune is equivariant and a universal approximator of equivariant functions. Additionally, we show that the method of \cite{kaba2022equivariance} used with appropriate loss functions, which we call \textit{equizero}, also gives excellent zero-shot and finetuned performance. Both equitune and equizero are special cases of $\lambda$-equitune. To show the simplicity and generality of our method, we validate on a wide range of diverse applications and models such as 1) image classification using CLIP, 2) deep Q-learning, 3) fairness in natural language generation (NLG), 4) compositional generalization in languages, and 5) image classification using pretrained CNNs such as Resnet and Alexnet.
Enhancing Protein Language Models with Structure-based Encoder and Pre-training
Mar 11, 2023Protein language models (PLMs) pre-trained on large-scale protein sequence corpora have achieved impressive performance on various downstream protein understanding tasks. Despite the ability to implicitly capture inter-residue contact information, transformer-based PLMs cannot encode protein structures explicitly for better structure-aware protein representations. Besides, the power of pre-training on available protein structures has not been explored for improving these PLMs, though structures are important to determine functions. To tackle these limitations, in this work, we enhance the PLMs with structure-based encoder and pre-training. We first explore feasible model architectures to combine the advantages of a state-of-the-art PLM (i.e., ESM-1b1) and a state-of-the-art protein structure encoder (i.e., GearNet). We empirically verify the ESM-GearNet that connects two encoders in a series way as the most effective combination model. To further improve the effectiveness of ESM-GearNet, we pre-train it on massive unlabeled protein structures with contrastive learning, which aligns representations of co-occurring subsequences so as to capture their biological correlation. Extensive experiments on EC and GO protein function prediction benchmarks demonstrate the superiority of ESM-GearNet over previous PLMs and structure encoders, and clear performance gains are further achieved by structure-based pre-training upon ESM-GearNet. Our implementation is available at https://github.com/DeepGraphLearning/GearNet.
Physics-Inspired Protein Encoder Pre-Training via Siamese Sequence-Structure Diffusion Trajectory Prediction
Jan 28, 2023Pre-training methods on proteins are recently gaining interest, leveraging either protein sequences or structures, while modeling their joint energy landscape is largely unexplored. In this work, inspired by the success of denoising diffusion models, we propose the DiffPreT approach to pre-train a protein encoder by sequence-structure multimodal diffusion modeling. DiffPreT guides the encoder to recover the native protein sequences and structures from the perturbed ones along the multimodal diffusion trajectory, which acquires the joint distribution of sequences and structures. Considering the essential protein conformational variations, we enhance DiffPreT by a physics-inspired method called Siamese Diffusion Trajectory Prediction (SiamDiff) to capture the correlation between different conformers of a protein. SiamDiff attains this goal by maximizing the mutual information between representations of diffusion trajectories of structurally-correlated conformers. We study the effectiveness of DiffPreT and SiamDiff on both atom- and residue-level structure-based protein understanding tasks. Experimental results show that the performance of DiffPreT is consistently competitive on all tasks, and SiamDiff achieves new state-of-the-art performance, considering the mean ranks on all tasks. The source code will be released upon acceptance.
Equi-Tuning: Group Equivariant Fine-Tuning of Pretrained Models
Oct 13, 2022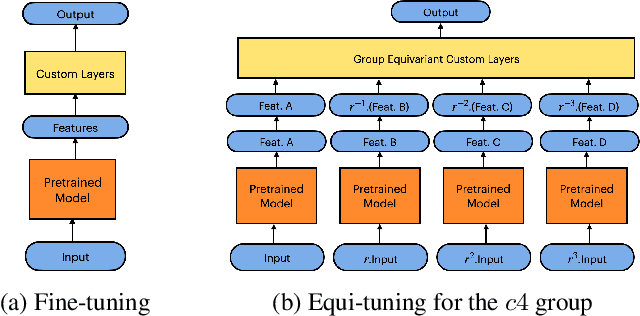
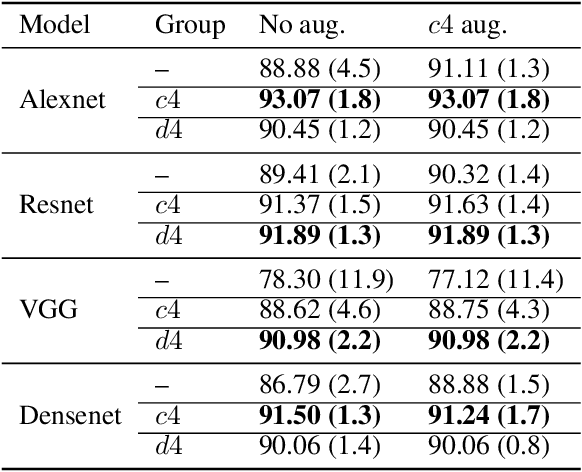

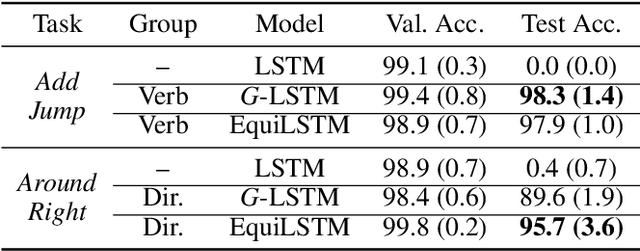
We introduce equi-tuning, a novel fine-tuning method that transforms (potentially non-equivariant) pretrained models into group equivariant models while incurring minimum $L_2$ loss between the feature representations of the pretrained and the equivariant models. Large pretrained models can be equi-tuned for different groups to satisfy the needs of various downstream tasks. Equi-tuned models benefit from both group equivariance as an inductive bias and semantic priors from pretrained models. We provide applications of equi-tuning on three different tasks: image classification, compositional generalization in language, and fairness in natural language generation (NLG). We also provide a novel group-theoretic definition for fairness in NLG. The effectiveness of this definition is shown by testing it against a standard empirical method of fairness in NLG. We provide experimental results for equi-tuning using a variety of pretrained models: Alexnet, Resnet, VGG, and Densenet for image classification; RNNs, GRUs, and LSTMs for compositional generalization; and GPT2 for fairness in NLG. We test these models on benchmark datasets across all considered tasks to show the generality and effectiveness of the proposed method.