 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMeasuring Scientific Capabilities of Language Models with a Systems Biology Dry Lab
Jul 02, 2025
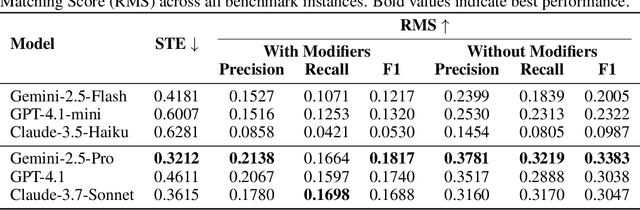
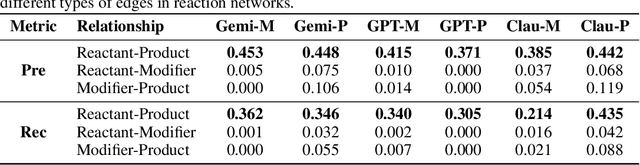
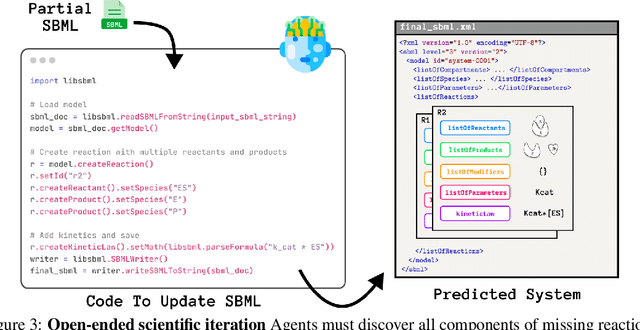
Designing experiments and result interpretations are core scientific competencies, particularly in biology, where researchers perturb complex systems to uncover the underlying systems. Recent efforts to evaluate the scientific capabilities of large language models (LLMs) fail to test these competencies because wet-lab experimentation is prohibitively expensive: in expertise, time and equipment. We introduce SciGym, a first-in-class benchmark that assesses LLMs' iterative experiment design and analysis abilities in open-ended scientific discovery tasks. SciGym overcomes the challenge of wet-lab costs by running a dry lab of biological systems. These models, encoded in Systems Biology Markup Language, are efficient for generating simulated data, making them ideal testbeds for experimentation on realistically complex systems. We evaluated six frontier LLMs on 137 small systems, and released a total of 350 systems. Our evaluation shows that while more capable models demonstrated superior performance, all models' performance declined significantly as system complexity increased, suggesting substantial room for improvement in the scientific capabilities of LLM agents.
Aligning Protein Conformation Ensemble Generation with Physical Feedback
May 30, 2025Protein dynamics play a crucial role in protein biological functions and properties, and their traditional study typically relies on time-consuming molecular dynamics (MD) simulations conducted in silico. Recent advances in generative modeling, particularly denoising diffusion models, have enabled efficient accurate protein structure prediction and conformation sampling by learning distributions over crystallographic structures. However, effectively integrating physical supervision into these data-driven approaches remains challenging, as standard energy-based objectives often lead to intractable optimization. In this paper, we introduce Energy-based Alignment (EBA), a method that aligns generative models with feedback from physical models, efficiently calibrating them to appropriately balance conformational states based on their energy differences. Experimental results on the MD ensemble benchmark demonstrate that EBA achieves state-of-the-art performance in generating high-quality protein ensembles. By improving the physical plausibility of generated structures, our approach enhances model predictions and holds promise for applications in structural biology and drug discovery.
Structure Language Models for Protein Conformation Generation
Oct 24, 2024
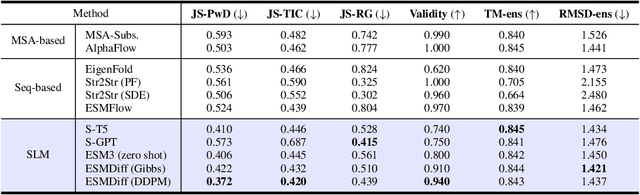
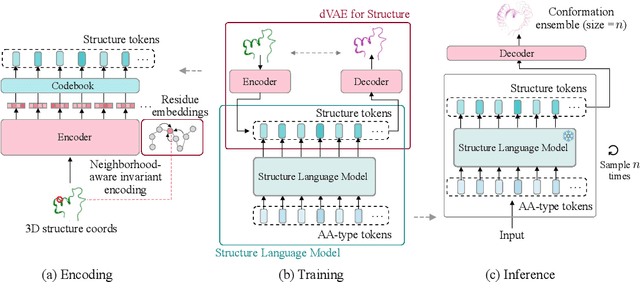
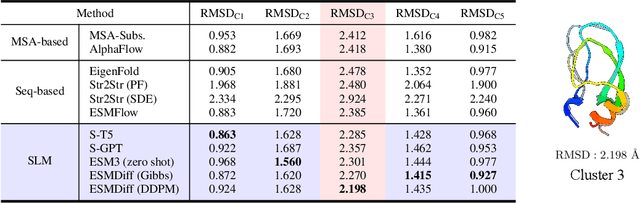
Proteins adopt multiple structural conformations to perform their diverse biological functions, and understanding these conformations is crucial for advancing drug discovery. Traditional physics-based simulation methods often struggle with sampling equilibrium conformations and are computationally expensive. Recently, deep generative models have shown promise in generating protein conformations as a more efficient alternative. However, these methods predominantly rely on the diffusion process within a 3D geometric space, which typically centers around the vicinity of metastable states and is often inefficient in terms of runtime. In this paper, we introduce Structure Language Modeling (SLM) as a novel framework for efficient protein conformation generation. Specifically, the protein structures are first encoded into a compact latent space using a discrete variational auto-encoder, followed by conditional language modeling that effectively captures sequence-specific conformation distributions. This enables a more efficient and interpretable exploration of diverse ensemble modes compared to existing methods. Based on this general framework, we instantiate SLM with various popular LM architectures as well as proposing the ESMDiff, a novel BERT-like structure language model fine-tuned from ESM3 with masked diffusion. We verify our approach in various scenarios, including the equilibrium dynamics of BPTI, conformational change pairs, and intrinsically disordered proteins. SLM provides a highly efficient solution, offering a 20-100x speedup than existing methods in generating diverse conformations, shedding light on promising avenues for future research.
Cell Morphology-Guided Small Molecule Generation with GFlowNets
Aug 09, 2024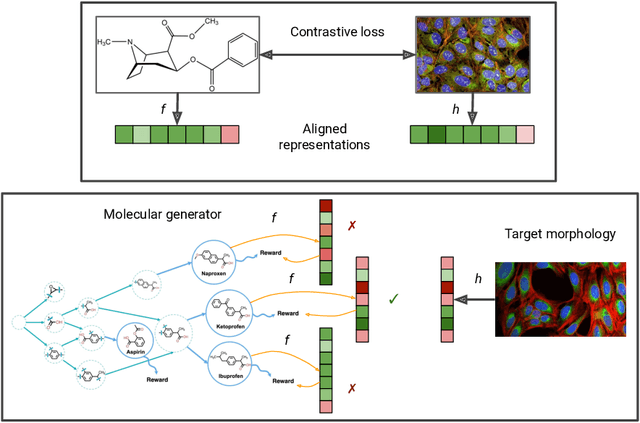


High-content phenotypic screening, including high-content imaging (HCI), has gained popularity in the last few years for its ability to characterize novel therapeutics without prior knowledge of the protein target. When combined with deep learning techniques to predict and represent molecular-phenotype interactions, these advancements hold the potential to significantly accelerate and enhance drug discovery applications. This work focuses on the novel task of HCI-guided molecular design. Generative models for molecule design could be guided by HCI data, for example with a supervised model that links molecules to phenotypes of interest as a reward function. However, limited labeled data, combined with the high-dimensional readouts, can make training these methods challenging and impractical. We consider an alternative approach in which we leverage an unsupervised multimodal joint embedding to define a latent similarity as a reward for GFlowNets. The proposed model learns to generate new molecules that could produce phenotypic effects similar to those of the given image target, without relying on pre-annotated phenotypic labels. We demonstrate that the proposed method generates molecules with high morphological and structural similarity to the target, increasing the likelihood of similar biological activity, as confirmed by an independent oracle model.
QGFN: Controllable Greediness with Action Values
Feb 07, 2024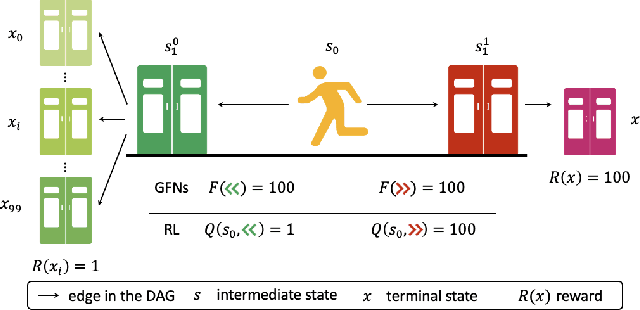

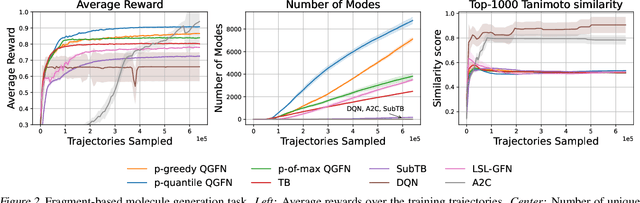
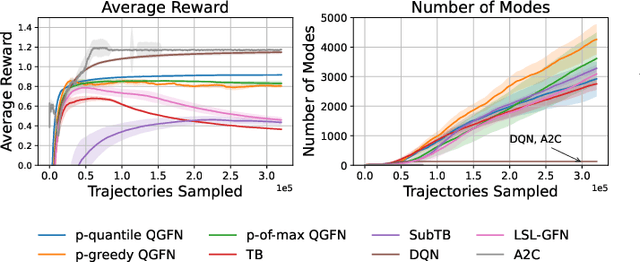
Generative Flow Networks (GFlowNets; GFNs) are a family of reward/energy-based generative methods for combinatorial objects, capable of generating diverse and high-utility samples. However, biasing GFNs towards producing high-utility samples is non-trivial. In this work, we leverage connections between GFNs and reinforcement learning (RL) and propose to combine the GFN policy with an action-value estimate, $Q$, to create greedier sampling policies which can be controlled by a mixing parameter. We show that several variants of the proposed method, QGFN, are able to improve on the number of high-reward samples generated in a variety of tasks without sacrificing diversity.