 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFrom Answers to States: Verifiable Process-Level Evaluation of Chemical Reasoning in Large Language Models
Jun 03, 2026Large language models are increasingly used as chemistry assistants, yet most chemistry benchmarks still score only final answers. This masks a critical failure mode: a model may output the correct molecule, product, or option while its reasoning violates chemical logic. Existing process-level evaluators are hard to scale because LLM judges and human step-level process annotation are costly, inconsistent, and vulnerable to hallucination. We introduce ChemCoTBench-V2, a rule-verifiable diagnostic benchmark for low-cost, auditable evaluation of structured, verifier-addressable chemical reasoning traces. It spans molecular understanding, molecule editing, molecular optimization, and reaction prediction, with 5,620 evaluation samples across 18 reporting tasks. Models must expose key intermediate steps in expert-designed templates, and those steps are checked with deterministic chemistry rules and, for closed-answer tasks, reference traces rather than another LLM judge. Open-ended molecular optimization is evaluated with oracle-verifiable state constraints rather than strict trace matching. The benchmark reports three separate signals: final-answer correctness, template adherence, and step-wise verifier correctness over expert-refined intermediate commitments. Experiments on frontier models reveal a persistent gap between final-answer success and structured-reasoning-state consistency: models often follow the requested format while failing chemical-step checks, or answer correctly with weak supporting reasoning. ChemCoTBench-V2 enables fine-grained model comparison and identifies the concrete step at which the trace first violates the verifier.
Plausibility Is Not Prediction: Contrastive Evidence for LLM-Based Cellular Perturbation Reasoning
May 31, 2026Perturbation experiments are central to understanding cellular mechanisms, but remain costly and sparse, motivating prediction of gene expression responses for unobserved conditions. A promising recent direction leverages large language models (LLMs) as "virtual cell" simulators-using stepwise, knowledge-grounded mechanistic reasoning to infer differential expression-pointing toward an interpretable, knowledge-driven paradigm that transcends purely data-driven approaches. However, we find that plausibility is not prediction: despite producing biologically plausible explanations, these methods fail to capture perturbation-specific effects: systematically overestimating differential expression, often underperforming a simple gene-frequency baseline in aggregate evaluations, and collapsing to chance-level performance at the per-gene level. This reveals a reliance on intrinsic gene response tendencies rather than true perturbation reasoning. We trace this failure to how evidence is presented: existing methods evaluate perturbation-gene pairs in isolation, without exposing how related perturbations differ in their effects on the same gene. To address this limitation, we introduce CORE (Contrastive Organization of Relational Evidence), which reframes prediction as a comparison task by organizing evidence into positive and negative outcomes from related perturbations. Using a biomedical knowledge graph for evidence retrieval, CORE improves calibration and substantially boosts perturbation-specific prediction in both LLM-based and non-LLM settings: for example, on drug-perturbation data, CORE-Reasoning improves Qwen3.5-9B aggregate metrics by up to 28.6%, while on generic perturbation data, CORE-Voting raises macro-per-gene AUROC from chance to 0.703 in average across four cell lines. This highlights contrastive evidence organization as essential to reliable LLM-based perturbation reasoning
Rigidity-Aware Geometric Pretraining for Protein Design and Conformational Ensembles
Mar 02, 2026Generative models have recently advanced $\textit{de novo}$ protein design by learning the statistical regularities of natural structures. However, current approaches face three key limitations: (1) Existing methods cannot jointly learn protein geometry and design tasks, where pretraining can be a solution; (2) Current pretraining methods mostly rely on local, non-rigid atomic representations for property prediction downstream tasks, limiting global geometric understanding for protein generation tasks; and (3) Existing approaches have yet to effectively model the rich dynamic and conformational information of protein structures. To overcome these issues, we introduce $\textbf{RigidSSL}$ ($\textit{Rigidity-Aware Self-Supervised Learning}$), a geometric pretraining framework that front-loads geometry learning prior to generative finetuning. Phase I (RigidSSL-Perturb) learns geometric priors from 432K structures from the AlphaFold Protein Structure Database with simulated perturbations. Phase II (RigidSSL-MD) refines these representations on 1.3K molecular dynamics trajectories to capture physically realistic transitions. Underpinning both phases is a bi-directional, rigidity-aware flow matching objective that jointly optimizes translational and rotational dynamics to maximize mutual information between conformations. Empirically, RigidSSL variants improve designability by up to 43\% while enhancing novelty and diversity in unconditional generation. Furthermore, RigidSSL-Perturb improves the success rate by 5.8\% in zero-shot motif scaffolding and RigidSSL-MD captures more biophysically realistic conformational ensembles in G protein-coupled receptor modeling. The code is available at: https://github.com/ZhanghanNi/RigidSSL.git.
PerturbDiff: Functional Diffusion for Single-Cell Perturbation Modeling
Feb 23, 2026Building Virtual Cells that can accurately simulate cellular responses to perturbations is a long-standing goal in systems biology. A fundamental challenge is that high-throughput single-cell sequencing is destructive: the same cell cannot be observed both before and after a perturbation. Thus, perturbation prediction requires mapping unpaired control and perturbed populations. Existing models address this by learning maps between distributions, but typically assume a single fixed response distribution when conditioned on observed cellular context (e.g., cell type) and the perturbation type. In reality, responses vary systematically due to unobservable latent factors such as microenvironmental fluctuations and complex batch effects, forming a manifold of possible distributions for the same observed conditions. To account for this variability, we introduce PerturbDiff, which shifts modeling from individual cells to entire distributions. By embedding distributions as points in a Hilbert space, we define a diffusion-based generative process operating directly over probability distributions. This allows PerturbDiff to capture population-level response shifts across hidden factors. Benchmarks on established datasets show that PerturbDiff achieves state-of-the-art performance in single-cell response prediction and generalizes substantially better to unseen perturbations. See our project page (https://katarinayuan.github.io/PerturbDiff-ProjectPage/), where code and data will be made publicly available (https://github.com/DeepGraphLearning/PerturbDiff).
InertialAR: Autoregressive 3D Molecule Generation with Inertial Frames
Oct 31, 2025Transformer-based autoregressive models have emerged as a unifying paradigm across modalities such as text and images, but their extension to 3D molecule generation remains underexplored. The gap stems from two fundamental challenges: (1) tokenizing molecules into a canonical 1D sequence of tokens that is invariant to both SE(3) transformations and atom index permutations, and (2) designing an architecture capable of modeling hybrid atom-based tokens that couple discrete atom types with continuous 3D coordinates. To address these challenges, we introduce InertialAR. InertialAR devises a canonical tokenization that aligns molecules to their inertial frames and reorders atoms to ensure SE(3) and permutation invariance. Moreover, InertialAR equips the attention mechanism with geometric awareness via geometric rotary positional encoding (GeoRoPE). In addition, it utilizes a hierarchical autoregressive paradigm to predict the next atom-based token, predicting the atom type first and then its 3D coordinates via Diffusion loss. Experimentally, InertialAR achieves state-of-the-art performance on 7 of the 10 evaluation metrics for unconditional molecule generation across QM9, GEOM-Drugs, and B3LYP. Moreover, it significantly outperforms strong baselines in controllable generation for targeted chemical functionality, attaining state-of-the-art results across all 5 metrics.
LacMaterial: Large Language Models as Analogical Chemists for Materials Discovery
Oct 25, 2025Analogical reasoning, the transfer of relational structures across contexts (e.g., planet is to sun as electron is to nucleus), is fundamental to scientific discovery. Yet human insight is often constrained by domain expertise and surface-level biases, limiting access to deeper, structure-driven analogies both within and across disciplines. Large language models (LLMs), trained on vast cross-domain data, present a promising yet underexplored tool for analogical reasoning in science. Here, we demonstrate that LLMs can generate novel battery materials by (1) retrieving cross-domain analogs and analogy-guided exemplars to steer exploration beyond conventional dopant substitutions, and (2) constructing in-domain analogical templates from few labeled examples to guide targeted exploitation. These explicit analogical reasoning strategies yield candidates outside established compositional spaces and outperform standard prompting baselines. Our findings position LLMs as interpretable, expert-like hypothesis generators that leverage analogy-driven generalization for scientific innovation.
UniFGVC: Universal Training-Free Few-Shot Fine-Grained Vision Classification via Attribute-Aware Multimodal Retrieval
Aug 06, 2025Few-shot fine-grained visual classification (FGVC) aims to leverage limited data to enable models to discriminate subtly distinct categories. Recent works mostly finetuned the pre-trained visual language models to achieve performance gain, yet suffering from overfitting and weak generalization. To deal with this, we introduce UniFGVC, a universal training-free framework that reformulates few-shot FGVC as multimodal retrieval. First, we propose the Category-Discriminative Visual Captioner (CDV-Captioner) to exploit the open-world knowledge of multimodal large language models (MLLMs) to generate a structured text description that captures the fine-grained attribute features distinguishing closely related classes. CDV-Captioner uses chain-of-thought prompting and visually similar reference images to reduce hallucination and enhance discrimination of generated captions. Using it we can convert each image into an image-description pair, enabling more comprehensive feature representation, and construct the multimodal category templates using few-shot samples for the subsequent retrieval pipeline. Then, off-the-shelf vision and text encoders embed query and template pairs, and FGVC is accomplished by retrieving the nearest template in the joint space. UniFGVC ensures broad compatibility with diverse MLLMs and encoders, offering reliable generalization and adaptability across few-shot FGVC scenarios. Extensive experiments on 12 FGVC benchmarks demonstrate its consistent superiority over prior few-shot CLIP-based methods and even several fully-supervised MLLMs-based approaches.
DisProtEdit: Exploring Disentangled Representations for Multi-Attribute Protein Editing
Jun 17, 2025We introduce DisProtEdit, a controllable protein editing framework that leverages dual-channel natural language supervision to learn disentangled representations of structural and functional properties. Unlike prior approaches that rely on joint holistic embeddings, DisProtEdit explicitly separates semantic factors, enabling modular and interpretable control. To support this, we construct SwissProtDis, a large-scale multimodal dataset where each protein sequence is paired with two textual descriptions, one for structure and one for function, automatically decomposed using a large language model. DisProtEdit aligns protein and text embeddings using alignment and uniformity objectives, while a disentanglement loss promotes independence between structural and functional semantics. At inference time, protein editing is performed by modifying one or both text inputs and decoding from the updated latent representation. Experiments on protein editing and representation learning benchmarks demonstrate that DisProtEdit performs competitively with existing methods while providing improved interpretability and controllability. On a newly constructed multi-attribute editing benchmark, the model achieves a both-hit success rate of up to 61.7%, highlighting its effectiveness in coordinating simultaneous structural and functional edits.
OBELiX: A Curated Dataset of Crystal Structures and Experimentally Measured Ionic Conductivities for Lithium Solid-State Electrolytes
Feb 20, 2025



Solid-state electrolyte batteries are expected to replace liquid electrolyte lithium-ion batteries in the near future thanks to their higher theoretical energy density and improved safety. However, their adoption is currently hindered by their lower effective ionic conductivity, a quantity that governs charge and discharge rates. Identifying highly ion-conductive materials using conventional theoretical calculations and experimental validation is both time-consuming and resource-intensive. While machine learning holds the promise to expedite this process, relevant ionic conductivity and structural data is scarce. Here, we present OBELiX, a domain-expert-curated database of $\sim$600 synthesized solid electrolyte materials and their experimentally measured room temperature ionic conductivities gathered from literature. Each material is described by their measured composition, space group and lattice parameters. A full-crystal description in the form of a crystallographic information file (CIF) is provided for ~320 structures for which atomic positions were available. We discuss various statistics and features of the dataset and provide training and testing splits that avoid data leakage. Finally, we benchmark seven existing ML models on the task of predicting ionic conductivity and discuss their performance. The goal of this work is to facilitate the use of machine learning for solid-state electrolyte materials discovery.
Baichuan-Omni-1.5 Technical Report
Jan 26, 2025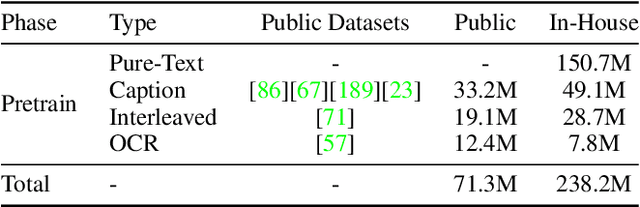
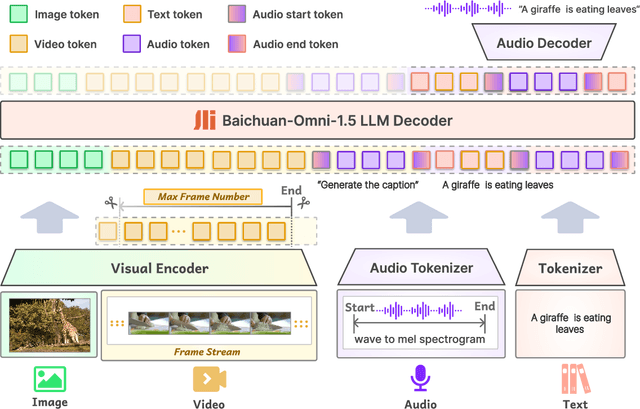
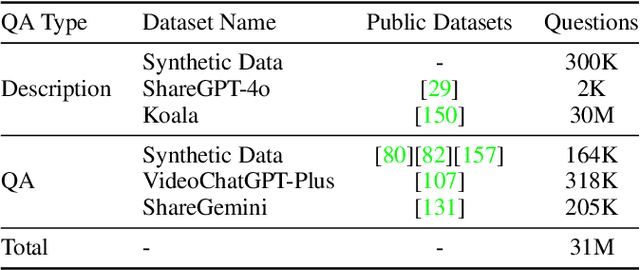
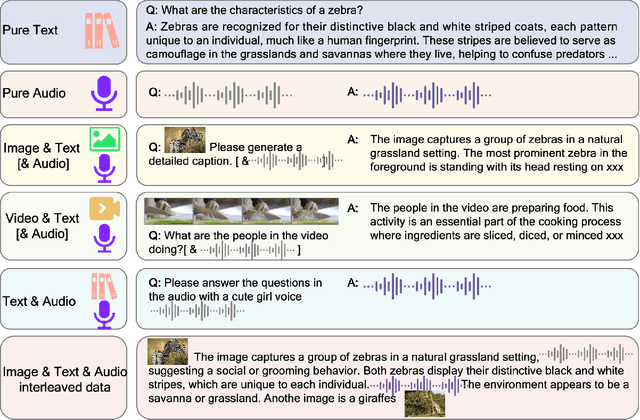
We introduce Baichuan-Omni-1.5, an omni-modal model that not only has omni-modal understanding capabilities but also provides end-to-end audio generation capabilities. To achieve fluent and high-quality interaction across modalities without compromising the capabilities of any modality, we prioritized optimizing three key aspects. First, we establish a comprehensive data cleaning and synthesis pipeline for multimodal data, obtaining about 500B high-quality data (text, audio, and vision). Second, an audio-tokenizer (Baichuan-Audio-Tokenizer) has been designed to capture both semantic and acoustic information from audio, enabling seamless integration and enhanced compatibility with MLLM. Lastly, we designed a multi-stage training strategy that progressively integrates multimodal alignment and multitask fine-tuning, ensuring effective synergy across all modalities. Baichuan-Omni-1.5 leads contemporary models (including GPT4o-mini and MiniCPM-o 2.6) in terms of comprehensive omni-modal capabilities. Notably, it achieves results comparable to leading models such as Qwen2-VL-72B across various multimodal medical benchmarks.