 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSmooth Dynamic Cutoffs for Machine Learning Interatomic Potentials
Jan 29, 2026Machine learning interatomic potentials (MLIPs) have proven to be wildly useful for molecular dynamics simulations, powering countless drug and materials discovery applications. However, MLIPs face two primary bottlenecks preventing them from reaching realistic simulation scales: inference time and memory consumption. In this work, we address both issues by challenging the long-held belief that the cutoff radius for the MLIP must be held to a fixed, constant value. For the first time, we introduce a dynamic cutoff formulation that still leads to stable, long timescale molecular dynamics simulation. In introducing the dynamic cutoff, we are able to induce sparsity onto the underlying atom graph by targeting a specific number of neighbors per atom, significantly reducing both memory consumption and inference time. We show the effectiveness of a dynamic cutoff by implementing it onto 4 state of the art MLIPs: MACE, Nequip, Orbv3, and TensorNet, leading to 2.26x less memory consumption and 2.04x faster inference time, depending on the model and atomic system. We also perform an extensive error analysis and find that the dynamic cutoff models exhibit minimal accuracy dropoff compared to their fixed cutoff counterparts on both materials and molecular datasets. All model implementations and training code will be fully open sourced.
Two Heads are Better than One: Distilling Large Language Model Features Into Small Models with Feature Decomposition and Mixture
Nov 11, 2025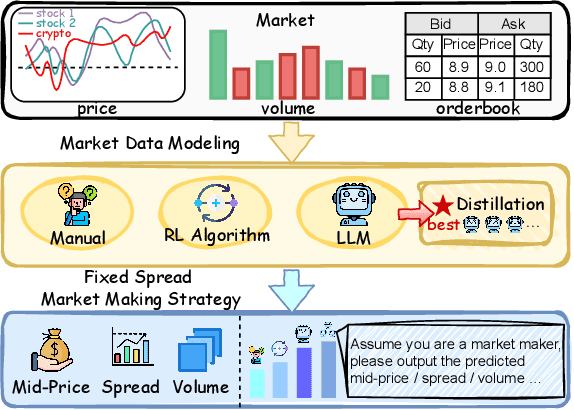
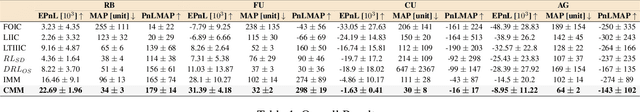
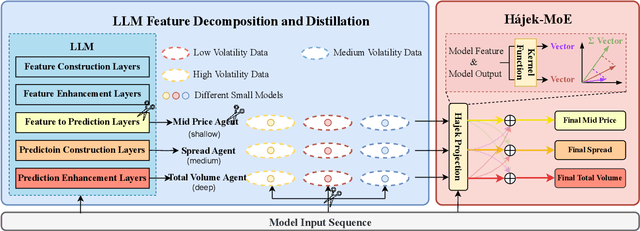

Market making (MM) through Reinforcement Learning (RL) has attracted significant attention in financial trading. With the development of Large Language Models (LLMs), more and more attempts are being made to apply LLMs to financial areas. A simple, direct application of LLM as an agent shows significant performance. Such methods are hindered by their slow inference speed, while most of the current research has not studied LLM distillation for this specific task. To address this, we first propose the normalized fluorescent probe to study the mechanism of the LLM's feature. Based on the observation found by our investigation, we propose Cooperative Market Making (CMM), a novel framework that decouples LLM features across three orthogonal dimensions: layer, task, and data. Various student models collaboratively learn simple LLM features along with different dimensions, with each model responsible for a distinct feature to achieve knowledge distillation. Furthermore, CMM introduces an Hájek-MoE to integrate the output of the student models by investigating the contribution of different models in a kernel function-generated common feature space. Extensive experimental results on four real-world market datasets demonstrate the superiority of CMM over the current distillation method and RL-based market-making strategies.
NeuralTouch: Neural Descriptors for Precise Sim-to-Real Tactile Robot Control
Oct 23, 2025Grasping accuracy is a critical prerequisite for precise object manipulation, often requiring careful alignment between the robot hand and object. Neural Descriptor Fields (NDF) offer a promising vision-based method to generate grasping poses that generalize across object categories. However, NDF alone can produce inaccurate poses due to imperfect camera calibration, incomplete point clouds, and object variability. Meanwhile, tactile sensing enables more precise contact, but existing approaches typically learn policies limited to simple, predefined contact geometries. In this work, we introduce NeuralTouch, a multimodal framework that integrates NDF and tactile sensing to enable accurate, generalizable grasping through gentle physical interaction. Our approach leverages NDF to implicitly represent the target contact geometry, from which a deep reinforcement learning (RL) policy is trained to refine the grasp using tactile feedback. This policy is conditioned on the neural descriptors and does not require explicit specification of contact types. We validate NeuralTouch through ablation studies in simulation and zero-shot transfer to real-world manipulation tasks--such as peg-out-in-hole and bottle lid opening--without additional fine-tuning. Results show that NeuralTouch significantly improves grasping accuracy and robustness over baseline methods, offering a general framework for precise, contact-rich robotic manipulation.
MIRA: Medical Time Series Foundation Model for Real-World Health Data
Jun 09, 2025
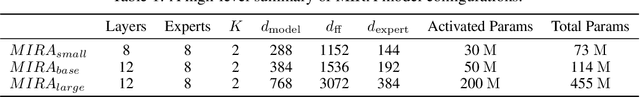

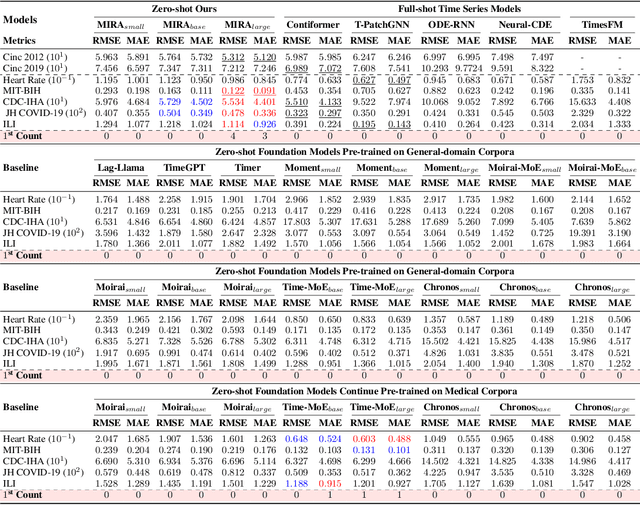
A unified foundation model for medical time series -- pretrained on open access and ethics board-approved medical corpora -- offers the potential to reduce annotation burdens, minimize model customization, and enable robust transfer across clinical institutions, modalities, and tasks, particularly in data-scarce or privacy-constrained environments. However, existing generalist time series foundation models struggle to handle medical time series data due to their inherent challenges, including irregular intervals, heterogeneous sampling rates, and frequent missing values. To address these challenges, we introduce MIRA, a unified foundation model specifically designed for medical time series forecasting. MIRA incorporates a Continuous-Time Rotary Positional Encoding that enables fine-grained modeling of variable time intervals, a frequency-specific mixture-of-experts layer that routes computation across latent frequency regimes to further promote temporal specialization, and a Continuous Dynamics Extrapolation Block based on Neural ODE that models the continuous trajectory of latent states, enabling accurate forecasting at arbitrary target timestamps. Pretrained on a large-scale and diverse medical corpus comprising over 454 billion time points collect from publicly available datasets, MIRA achieves reductions in forecasting errors by an average of 10% and 7% in out-of-distribution and in-distribution scenarios, respectively, when compared to other zero-shot and fine-tuned baselines. We also introduce a comprehensive benchmark spanning multiple downstream clinical tasks, establishing a foundation for future research in medical time series modeling.
Two-way Evidence self-Alignment based Dual-Gated Reasoning Enhancement
May 22, 2025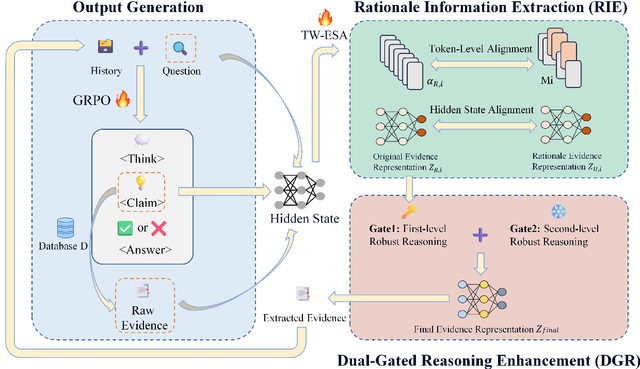
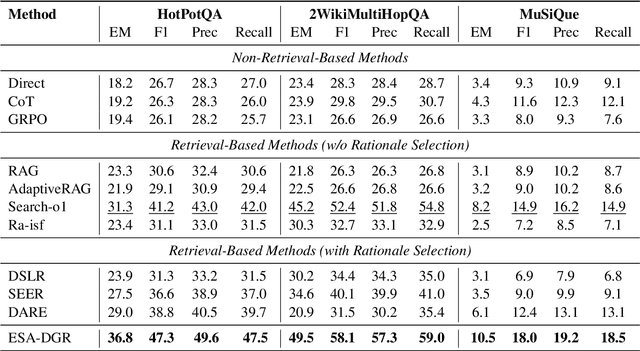
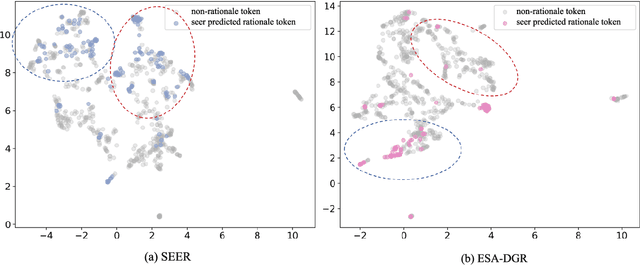
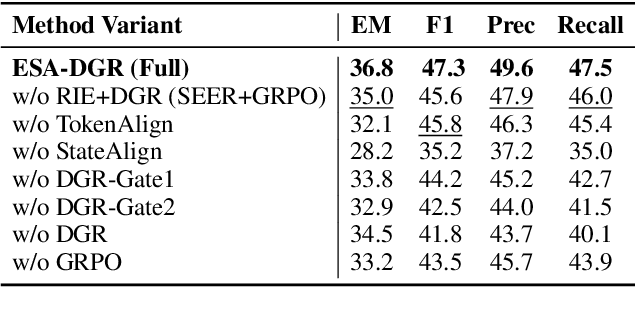
Large language models (LLMs) encounter difficulties in knowledge-intensive multi-step reasoning (KIMSR) tasks. One challenge is how to effectively extract and represent rationale evidence. The current methods often extract semantically relevant but logically irrelevant evidence, resulting in flawed reasoning and inaccurate responses. We propose a two-way evidence self-alignment (TW-ESA) module, which utilizes the mutual alignment between strict reasoning and LLM reasoning to enhance its understanding of the causal logic of evidence, thereby addressing the first challenge. Another challenge is how to utilize the rationale evidence and LLM's intrinsic knowledge for accurate reasoning when the evidence contains uncertainty. We propose a dual-gated reasoning enhancement (DGR) module to gradually fuse useful knowledge of LLM within strict reasoning, which can enable the model to perform accurate reasoning by focusing on causal elements in the evidence and exhibit greater robustness. The two modules are collaboratively trained in a unified framework ESA-DGR. Extensive experiments on three diverse and challenging KIMSR datasets reveal that ESA-DGR significantly surpasses state-of-the-art LLM-based fine-tuning methods, with remarkable average improvements of 4% in exact match (EM) and 5% in F1 score. The implementation code is available at https://anonymous.4open.science/r/ESA-DGR-2BF8.
TarDiff: Target-Oriented Diffusion Guidance for Synthetic Electronic Health Record Time Series Generation
Apr 24, 2025Synthetic Electronic Health Record (EHR) time-series generation is crucial for advancing clinical machine learning models, as it helps address data scarcity by providing more training data. However, most existing approaches focus primarily on replicating statistical distributions and temporal dependencies of real-world data. We argue that fidelity to observed data alone does not guarantee better model performance, as common patterns may dominate, limiting the representation of rare but important conditions. This highlights the need for generate synthetic samples to improve performance of specific clinical models to fulfill their target outcomes. To address this, we propose TarDiff, a novel target-oriented diffusion framework that integrates task-specific influence guidance into the synthetic data generation process. Unlike conventional approaches that mimic training data distributions, TarDiff optimizes synthetic samples by quantifying their expected contribution to improving downstream model performance through influence functions. Specifically, we measure the reduction in task-specific loss induced by synthetic samples and embed this influence gradient into the reverse diffusion process, thereby steering the generation towards utility-optimized data. Evaluated on six publicly available EHR datasets, TarDiff achieves state-of-the-art performance, outperforming existing methods by up to 20.4% in AUPRC and 18.4% in AUROC. Our results demonstrate that TarDiff not only preserves temporal fidelity but also enhances downstream model performance, offering a robust solution to data scarcity and class imbalance in healthcare analytics.
Can Competition Enhance the Proficiency of Agents Powered by Large Language Models in the Realm of News-driven Time Series Forecasting?
Apr 14, 2025Multi-agents-based news-driven time series forecasting is considered as a potential paradigm shift in the era of large language models (LLMs). The challenge of this task lies in measuring the influences of different news events towards the fluctuations of time series. This requires agents to possess stronger abilities of innovative thinking and the identifying misleading logic. However, the existing multi-agent discussion framework has limited enhancement on time series prediction in terms of optimizing these two capabilities. Inspired by the role of competition in fostering innovation, this study embeds a competition mechanism within the multi-agent discussion to enhance agents' capability of generating innovative thoughts. Furthermore, to bolster the model's proficiency in identifying misleading information, we incorporate a fine-tuned small-scale LLM model within the reflective stage, offering auxiliary decision-making support. Experimental results confirm that the competition can boost agents' capacity for innovative thinking, which can significantly improve the performances of time series prediction. Similar to the findings of social science, the intensity of competition within this framework can influence the performances of agents, providing a new perspective for studying LLMs-based multi-agent systems.
Cross-functional transferability in universal machine learning interatomic potentials
Apr 07, 2025The rapid development of universal machine learning interatomic potentials (uMLIPs) has demonstrated the possibility for generalizable learning of the universal potential energy surface. In principle, the accuracy of uMLIPs can be further improved by bridging the model from lower-fidelity datasets to high-fidelity ones. In this work, we analyze the challenge of this transfer learning problem within the CHGNet framework. We show that significant energy scale shifts and poor correlations between GGA and r$^2$SCAN pose challenges to cross-functional data transferability in uMLIPs. By benchmarking different transfer learning approaches on the MP-r$^2$SCAN dataset of 0.24 million structures, we demonstrate the importance of elemental energy referencing in the transfer learning of uMLIPs. By comparing the scaling law with and without the pre-training on a low-fidelity dataset, we show that significant data efficiency can still be achieved through transfer learning, even with a target dataset of sub-million structures. We highlight the importance of proper transfer learning and multi-fidelity learning in creating next-generation uMLIPs on high-fidelity data.
Structuring Scientific Innovation: A Framework for Modeling and Discovering Impactful Knowledge Combinations
Mar 25, 2025The emergence of large language models offers new possibilities for structured exploration of scientific knowledge. Rather than viewing scientific discovery as isolated ideas or content, we propose a structured approach that emphasizes the role of method combinations in shaping disruptive insights. Specifically, we investigate how knowledge unit--especially those tied to methodological design--can be modeled and recombined to yield research breakthroughs. Our proposed framework addresses two key challenges. First, we introduce a contrastive learning-based mechanism to identify distinguishing features of historically disruptive method combinations within problem-driven contexts. Second, we propose a reasoning-guided Monte Carlo search algorithm that leverages the chain-of-thought capability of LLMs to identify promising knowledge recombinations for new problem statements.Empirical studies across multiple domains show that the framework is capable of modeling the structural dynamics of innovation and successfully highlights combinations with high disruptive potential. This research provides a new path for computationally guided scientific ideation grounded in structured reasoning and historical data modeling.
A practical guide to machine learning interatomic potentials -- Status and future
Mar 12, 2025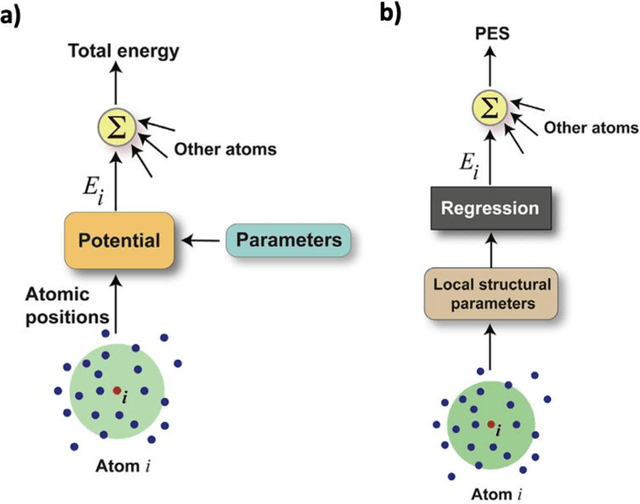

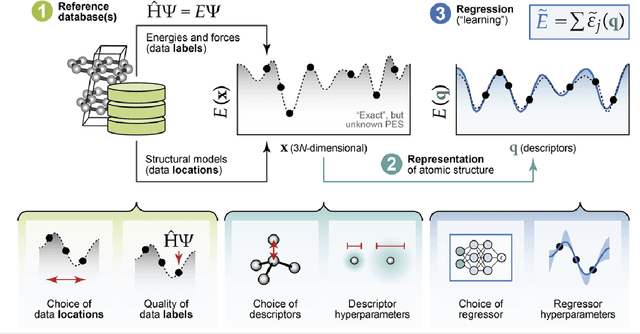
The rapid development and large body of literature on machine learning interatomic potentials (MLIPs) can make it difficult to know how to proceed for researchers who are not experts but wish to use these tools. The spirit of this review is to help such researchers by serving as a practical, accessible guide to the state-of-the-art in MLIPs. This review paper covers a broad range of topics related to MLIPs, including (i) central aspects of how and why MLIPs are enablers of many exciting advancements in molecular modeling, (ii) the main underpinnings of different types of MLIPs, including their basic structure and formalism, (iii) the potentially transformative impact of universal MLIPs for both organic and inorganic systems, including an overview of the most recent advances, capabilities, downsides, and potential applications of this nascent class of MLIPs, (iv) a practical guide for estimating and understanding the execution speed of MLIPs, including guidance for users based on hardware availability, type of MLIP used, and prospective simulation size and time, (v) a manual for what MLIP a user should choose for a given application by considering hardware resources, speed requirements, energy and force accuracy requirements, as well as guidance for choosing pre-trained potentials or fitting a new potential from scratch, (vi) discussion around MLIP infrastructure, including sources of training data, pre-trained potentials, and hardware resources for training, (vii) summary of some key limitations of present MLIPs and current approaches to mitigate such limitations, including methods of including long-range interactions, handling magnetic systems, and treatment of excited states, and finally (viii) we finish with some more speculative thoughts on what the future holds for the development and application of MLIPs over the next 3-10+ years.