 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFrom Knowledge to Action: Outcomes of the 2025 Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry
May 04, 2026Large language models (LLMs) are rapidly changing how researchers in materials science and chemistry discover, organize, and act on scientific knowledge. This paper analyzes a broad set of community-developed LLM applications in an effort to identify emerging patterns in how these systems can be used across the scientific research lifecycle. We organize the projects into two complementary categories: Knowledge Infrastructure, systems that structure, retrieve, synthesize, and validate scientific information; and Action Systems, systems that execute, coordinate, or automate scientific work across computational and experimental environments. The submissions reveal a shift from single-purpose LLM tools toward integrated, multi-agent workflows that combine retrieval, reasoning, tool use, and domain-specific validation. Prominent themes include retrieval-augmented generation as grounding infrastructure, persistent structured knowledge representations, multimodal and multilingual scientific inputs, and early progress toward laboratory-integrated closed-loop systems. Together, these results suggest that LLMs are evolving from general-purpose assistants into composable infrastructure for scientific reasoning and action. This work provides a community snapshot of that transition and a practical taxonomy for understanding emerging LLM-enabled workflows in materials science and chemistry.
34 Examples of LLM Applications in Materials Science and Chemistry: Towards Automation, Assistants, Agents, and Accelerated Scientific Discovery
May 05, 2025
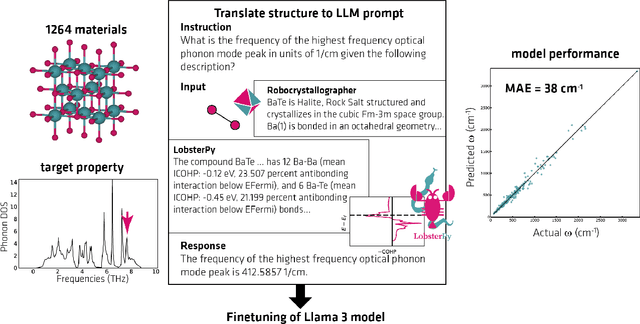
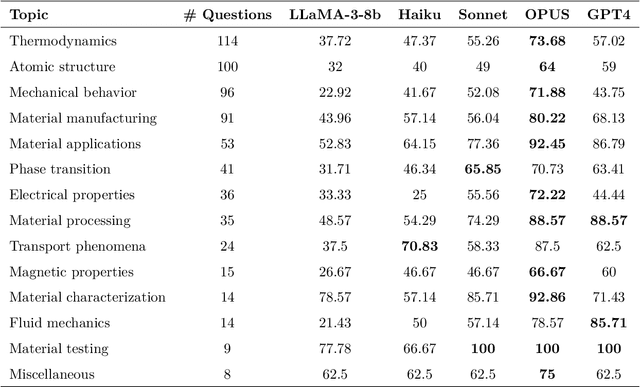
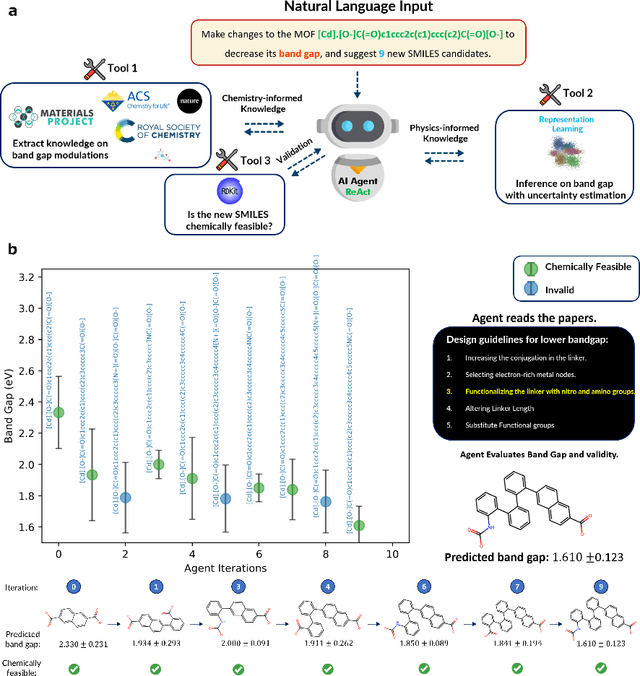
Large Language Models (LLMs) are reshaping many aspects of materials science and chemistry research, enabling advances in molecular property prediction, materials design, scientific automation, knowledge extraction, and more. Recent developments demonstrate that the latest class of models are able to integrate structured and unstructured data, assist in hypothesis generation, and streamline research workflows. To explore the frontier of LLM capabilities across the research lifecycle, we review applications of LLMs through 34 total projects developed during the second annual Large Language Model Hackathon for Applications in Materials Science and Chemistry, a global hybrid event. These projects spanned seven key research areas: (1) molecular and material property prediction, (2) molecular and material design, (3) automation and novel interfaces, (4) scientific communication and education, (5) research data management and automation, (6) hypothesis generation and evaluation, and (7) knowledge extraction and reasoning from the scientific literature. Collectively, these applications illustrate how LLMs serve as versatile predictive models, platforms for rapid prototyping of domain-specific tools, and much more. In particular, improvements in both open source and proprietary LLM performance through the addition of reasoning, additional training data, and new techniques have expanded effectiveness, particularly in low-data environments and interdisciplinary research. As LLMs continue to improve, their integration into scientific workflows presents both new opportunities and new challenges, requiring ongoing exploration, continued refinement, and further research to address reliability, interpretability, and reproducibility.
A practical guide to machine learning interatomic potentials -- Status and future
Mar 12, 2025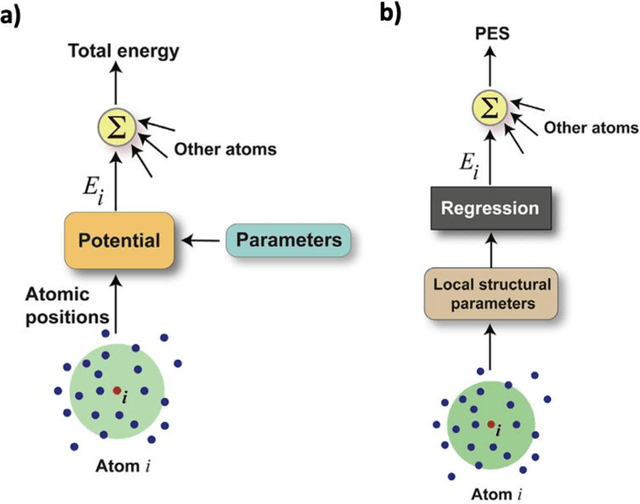

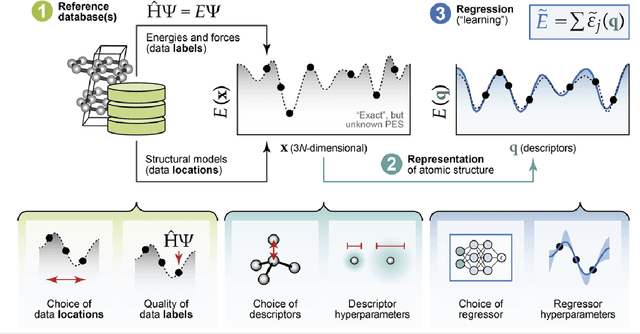
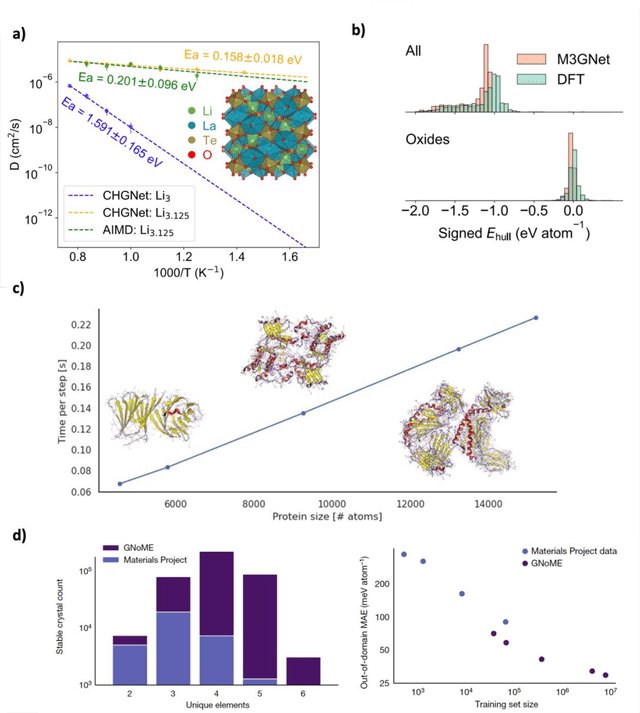
The rapid development and large body of literature on machine learning interatomic potentials (MLIPs) can make it difficult to know how to proceed for researchers who are not experts but wish to use these tools. The spirit of this review is to help such researchers by serving as a practical, accessible guide to the state-of-the-art in MLIPs. This review paper covers a broad range of topics related to MLIPs, including (i) central aspects of how and why MLIPs are enablers of many exciting advancements in molecular modeling, (ii) the main underpinnings of different types of MLIPs, including their basic structure and formalism, (iii) the potentially transformative impact of universal MLIPs for both organic and inorganic systems, including an overview of the most recent advances, capabilities, downsides, and potential applications of this nascent class of MLIPs, (iv) a practical guide for estimating and understanding the execution speed of MLIPs, including guidance for users based on hardware availability, type of MLIP used, and prospective simulation size and time, (v) a manual for what MLIP a user should choose for a given application by considering hardware resources, speed requirements, energy and force accuracy requirements, as well as guidance for choosing pre-trained potentials or fitting a new potential from scratch, (vi) discussion around MLIP infrastructure, including sources of training data, pre-trained potentials, and hardware resources for training, (vii) summary of some key limitations of present MLIPs and current approaches to mitigate such limitations, including methods of including long-range interactions, handling magnetic systems, and treatment of excited states, and finally (viii) we finish with some more speculative thoughts on what the future holds for the development and application of MLIPs over the next 3-10+ years.
Reflections from the 2024 Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry
Nov 20, 2024
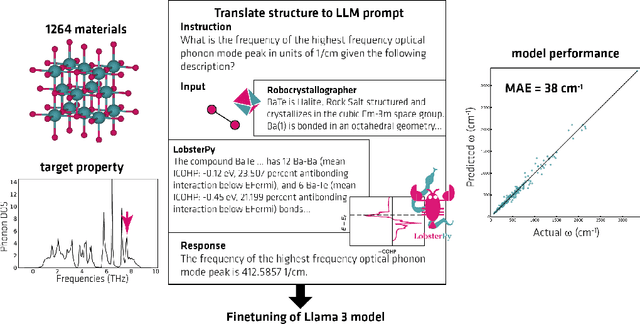
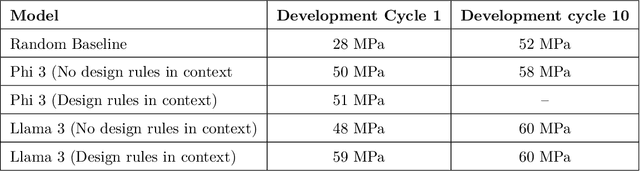
Here, we present the outcomes from the second Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry, which engaged participants across global hybrid locations, resulting in 34 team submissions. The submissions spanned seven key application areas and demonstrated the diverse utility of LLMs for applications in (1) molecular and material property prediction; (2) molecular and material design; (3) automation and novel interfaces; (4) scientific communication and education; (5) research data management and automation; (6) hypothesis generation and evaluation; and (7) knowledge extraction and reasoning from scientific literature. Each team submission is presented in a summary table with links to the code and as brief papers in the appendix. Beyond team results, we discuss the hackathon event and its hybrid format, which included physical hubs in Toronto, Montreal, San Francisco, Berlin, Lausanne, and Tokyo, alongside a global online hub to enable local and virtual collaboration. Overall, the event highlighted significant improvements in LLM capabilities since the previous year's hackathon, suggesting continued expansion of LLMs for applications in materials science and chemistry research. These outcomes demonstrate the dual utility of LLMs as both multipurpose models for diverse machine learning tasks and platforms for rapid prototyping custom applications in scientific research.
Twins in rotational spectroscopy: Does a rotational spectrum uniquely identify a molecule?
Apr 05, 2024


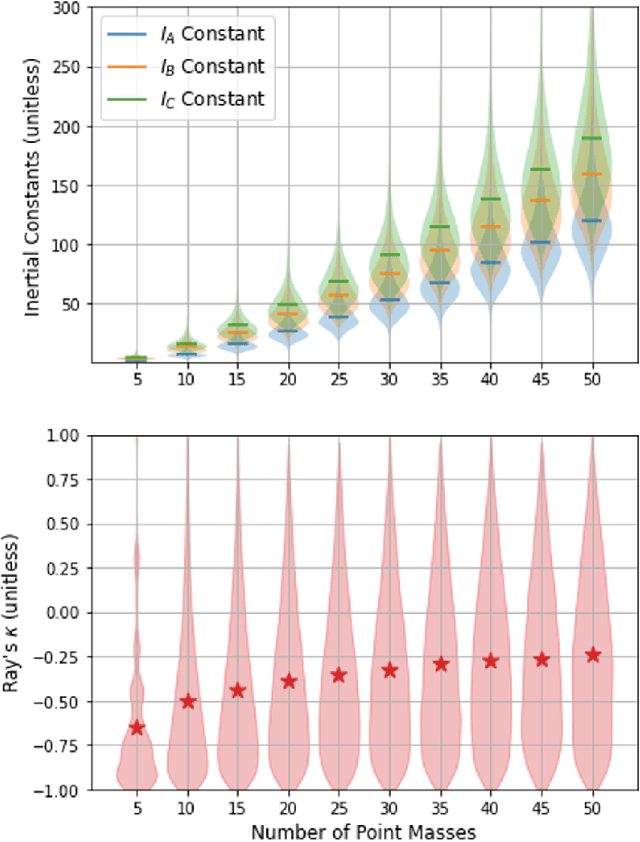
Rotational spectroscopy is the most accurate method for determining structures of molecules in the gas phase. It is often assumed that a rotational spectrum is a unique "fingerprint" of a molecule. The availability of large molecular databases and the development of artificial intelligence methods for spectroscopy makes the testing of this assumption timely. In this paper, we pose the determination of molecular structures from rotational spectra as an inverse problem. Within this framework, we adopt a funnel-based approach to search for molecular twins, which are two or more molecules, which have similar rotational spectra but distinctly different molecular structures. We demonstrate that there are twins within standard levels of computational accuracy by generating rotational constants for many molecules from several large molecular databases, indicating the inverse problem is ill-posed. However, some twins can be distinguished by increasing the accuracy of the theoretical methods or by performing additional experiments.
Trillion Parameter AI Serving Infrastructure for Scientific Discovery: A Survey and Vision
Feb 05, 2024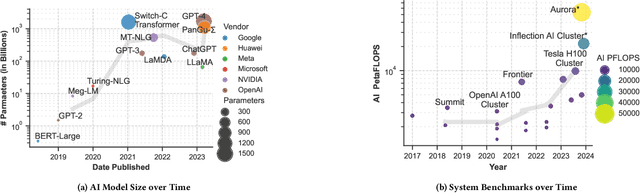
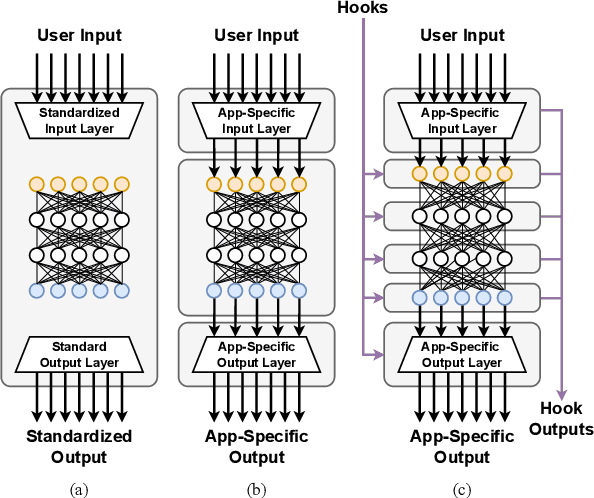
Deep learning methods are transforming research, enabling new techniques, and ultimately leading to new discoveries. As the demand for more capable AI models continues to grow, we are now entering an era of Trillion Parameter Models (TPM), or models with more than a trillion parameters -- such as Huawei's PanGu-$\Sigma$. We describe a vision for the ecosystem of TPM users and providers that caters to the specific needs of the scientific community. We then outline the significant technical challenges and open problems in system design for serving TPMs to enable scientific research and discovery. Specifically, we describe the requirements of a comprehensive software stack and interfaces to support the diverse and flexible requirements of researchers.
Accelerating Electronic Stopping Power Predictions by 10 Million Times with a Combination of Time-Dependent Density Functional Theory and Machine Learning
Nov 01, 2023Knowing the rate at which particle radiation releases energy in a material, the stopping power, is key to designing nuclear reactors, medical treatments, semiconductor and quantum materials, and many other technologies. While the nuclear contribution to stopping power, i.e., elastic scattering between atoms, is well understood in the literature, the route for gathering data on the electronic contribution has for decades remained costly and reliant on many simplifying assumptions, including that materials are isotropic. We establish a method that combines time-dependent density functional theory (TDDFT) and machine learning to reduce the time to assess new materials to mere hours on a supercomputer and provides valuable data on how atomic details influence electronic stopping. Our approach uses TDDFT to compute the electronic stopping contributions to stopping power from first principles in several directions and then machine learning to interpolate to other directions at rates 10 million times higher. We demonstrate the combined approach in a study of proton irradiation in aluminum and employ it to predict how the depth of maximum energy deposition, the "Bragg Peak," varies depending on incident angle -- a quantity otherwise inaccessible to modelers. The lack of any experimental information requirement makes our method applicable to most materials, and its speed makes it a prime candidate for enabling quantum-to-continuum models of radiation damage. The prospect of reusing valuable TDDFT data for training the model make our approach appealing for applications in the age of materials data science.
Towards a Modular Architecture for Science Factories
Aug 18, 2023Advances in robotic automation, high-performance computing (HPC), and artificial intelligence (AI) encourage us to conceive of science factories: large, general-purpose computation- and AI-enabled self-driving laboratories (SDLs) with the generality and scale needed both to tackle large discovery problems and to support thousands of scientists. Science factories require modular hardware and software that can be replicated for scale and (re)configured to support many applications. To this end, we propose a prototype modular science factory architecture in which reconfigurable modules encapsulating scientific instruments are linked with manipulators to form workcells, that can themselves be combined to form larger assemblages, and linked with distributed computing for simulation, AI model training and inference, and related tasks. Workflows that perform sets of actions on modules can be specified, and various applications, comprising workflows plus associated computational and data manipulation steps, can be run concurrently. We report on our experiences prototyping this architecture and applying it in experiments involving 15 different robotic apparatus, five applications (one in education, two in biology, two in materials), and a variety of workflows, across four laboratories. We describe the reuse of modules, workcells, and workflows in different applications, the migration of applications between workcells, and the use of digital twins, and suggest directions for future work aimed at yet more generality and scalability. Code and data are available at https://ad-sdl.github.io/wei2023 and in the Supplementary Information
14 Examples of How LLMs Can Transform Materials Science and Chemistry: A Reflection on a Large Language Model Hackathon
Jun 13, 2023



Chemistry and materials science are complex. Recently, there have been great successes in addressing this complexity using data-driven or computational techniques. Yet, the necessity of input structured in very specific forms and the fact that there is an ever-growing number of tools creates usability and accessibility challenges. Coupled with the reality that much data in these disciplines is unstructured, the effectiveness of these tools is limited. Motivated by recent works that indicated that large language models (LLMs) might help address some of these issues, we organized a hackathon event on the applications of LLMs in chemistry, materials science, and beyond. This article chronicles the projects built as part of this hackathon. Participants employed LLMs for various applications, including predicting properties of molecules and materials, designing novel interfaces for tools, extracting knowledge from unstructured data, and developing new educational applications. The diverse topics and the fact that working prototypes could be generated in less than two days highlight that LLMs will profoundly impact the future of our fields. The rich collection of ideas and projects also indicates that the applications of LLMs are not limited to materials science and chemistry but offer potential benefits to a wide range of scientific disciplines.
Deep Learning for Automated Experimentation in Scanning Transmission Electron Microscopy
Apr 04, 2023



Machine learning (ML) has become critical for post-acquisition data analysis in (scanning) transmission electron microscopy, (S)TEM, imaging and spectroscopy. An emerging trend is the transition to real-time analysis and closed-loop microscope operation. The effective use of ML in electron microscopy now requires the development of strategies for microscopy-centered experiment workflow design and optimization. Here, we discuss the associated challenges with the transition to active ML, including sequential data analysis and out-of-distribution drift effects, the requirements for the edge operation, local and cloud data storage, and theory in the loop operations. Specifically, we discuss the relative contributions of human scientists and ML agents in the ideation, orchestration, and execution of experimental workflows and the need to develop universal hyper languages that can apply across multiple platforms. These considerations will collectively inform the operationalization of ML in next-generation experimentation.