 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAdvances and Challenges in Foundation Agents: From Brain-Inspired Intelligence to Evolutionary, Collaborative, and Safe Systems
Mar 31, 2025The advent of large language models (LLMs) has catalyzed a transformative shift in artificial intelligence, paving the way for advanced intelligent agents capable of sophisticated reasoning, robust perception, and versatile action across diverse domains. As these agents increasingly drive AI research and practical applications, their design, evaluation, and continuous improvement present intricate, multifaceted challenges. This survey provides a comprehensive overview, framing intelligent agents within a modular, brain-inspired architecture that integrates principles from cognitive science, neuroscience, and computational research. We structure our exploration into four interconnected parts. First, we delve into the modular foundation of intelligent agents, systematically mapping their cognitive, perceptual, and operational modules onto analogous human brain functionalities, and elucidating core components such as memory, world modeling, reward processing, and emotion-like systems. Second, we discuss self-enhancement and adaptive evolution mechanisms, exploring how agents autonomously refine their capabilities, adapt to dynamic environments, and achieve continual learning through automated optimization paradigms, including emerging AutoML and LLM-driven optimization strategies. Third, we examine collaborative and evolutionary multi-agent systems, investigating the collective intelligence emerging from agent interactions, cooperation, and societal structures, highlighting parallels to human social dynamics. Finally, we address the critical imperative of building safe, secure, and beneficial AI systems, emphasizing intrinsic and extrinsic security threats, ethical alignment, robustness, and practical mitigation strategies necessary for trustworthy real-world deployment.
MOFA: Discovering Materials for Carbon Capture with a GenAI- and Simulation-Based Workflow
Jan 18, 2025We present MOFA, an open-source generative AI (GenAI) plus simulation workflow for high-throughput generation of metal-organic frameworks (MOFs) on large-scale high-performance computing (HPC) systems. MOFA addresses key challenges in integrating GPU-accelerated computing for GPU-intensive GenAI tasks, including distributed training and inference, alongside CPU- and GPU-optimized tasks for screening and filtering AI-generated MOFs using molecular dynamics, density functional theory, and Monte Carlo simulations. These heterogeneous tasks are unified within an online learning framework that optimizes the utilization of available CPU and GPU resources across HPC systems. Performance metrics from a 450-node (14,400 AMD Zen 3 CPUs + 1800 NVIDIA A100 GPUs) supercomputer run demonstrate that MOFA achieves high-throughput generation of novel MOF structures, with CO$_2$ adsorption capacities ranking among the top 10 in the hypothetical MOF (hMOF) dataset. Furthermore, the production of high-quality MOFs exhibits a linear relationship with the number of nodes utilized. The modular architecture of MOFA will facilitate its integration into other scientific applications that dynamically combine GenAI with large-scale simulations.
Employing Artificial Intelligence to Steer Exascale Workflows with Colmena
Aug 26, 2024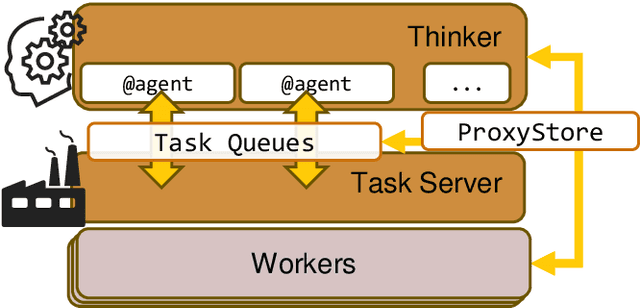
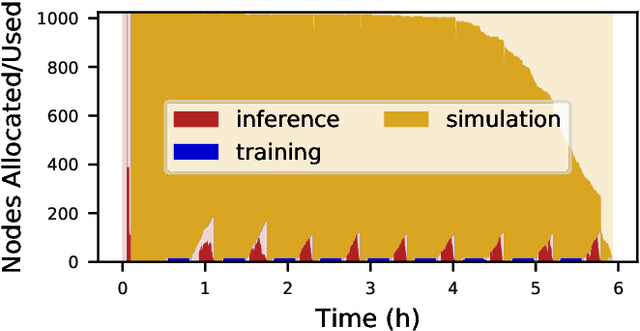
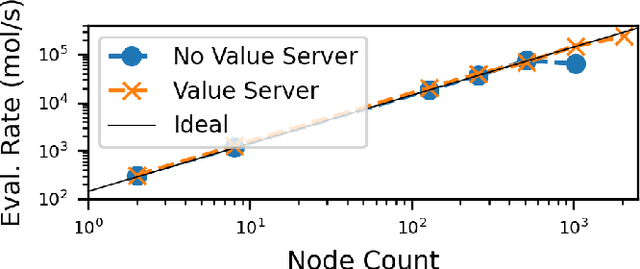
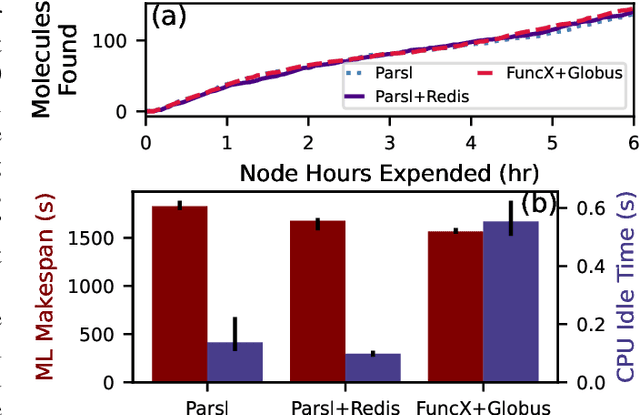
Computational workflows are a common class of application on supercomputers, yet the loosely coupled and heterogeneous nature of workflows often fails to take full advantage of their capabilities. We created Colmena to leverage the massive parallelism of a supercomputer by using Artificial Intelligence (AI) to learn from and adapt a workflow as it executes. Colmena allows scientists to define how their application should respond to events (e.g., task completion) as a series of cooperative agents. In this paper, we describe the design of Colmena, the challenges we overcame while deploying applications on exascale systems, and the science workflows we have enhanced through interweaving AI. The scaling challenges we discuss include developing steering strategies that maximize node utilization, introducing data fabrics that reduce communication overhead of data-intensive tasks, and implementing workflow tasks that cache costly operations between invocations. These innovations coupled with a variety of application patterns accessible through our agent-based steering model have enabled science advances in chemistry, biophysics, and materials science using different types of AI. Our vision is that Colmena will spur creative solutions that harness AI across many domains of scientific computing.
Trillion Parameter AI Serving Infrastructure for Scientific Discovery: A Survey and Vision
Feb 05, 2024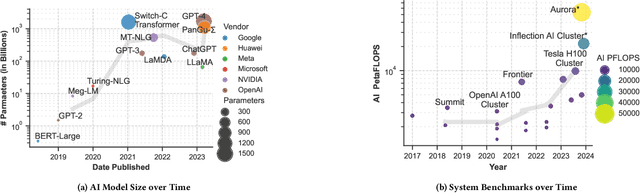
Deep learning methods are transforming research, enabling new techniques, and ultimately leading to new discoveries. As the demand for more capable AI models continues to grow, we are now entering an era of Trillion Parameter Models (TPM), or models with more than a trillion parameters -- such as Huawei's PanGu-$\Sigma$. We describe a vision for the ecosystem of TPM users and providers that caters to the specific needs of the scientific community. We then outline the significant technical challenges and open problems in system design for serving TPMs to enable scientific research and discovery. Specifically, we describe the requirements of a comprehensive software stack and interfaces to support the diverse and flexible requirements of researchers.
Accelerating Electronic Stopping Power Predictions by 10 Million Times with a Combination of Time-Dependent Density Functional Theory and Machine Learning
Nov 01, 2023Knowing the rate at which particle radiation releases energy in a material, the stopping power, is key to designing nuclear reactors, medical treatments, semiconductor and quantum materials, and many other technologies. While the nuclear contribution to stopping power, i.e., elastic scattering between atoms, is well understood in the literature, the route for gathering data on the electronic contribution has for decades remained costly and reliant on many simplifying assumptions, including that materials are isotropic. We establish a method that combines time-dependent density functional theory (TDDFT) and machine learning to reduce the time to assess new materials to mere hours on a supercomputer and provides valuable data on how atomic details influence electronic stopping. Our approach uses TDDFT to compute the electronic stopping contributions to stopping power from first principles in several directions and then machine learning to interpolate to other directions at rates 10 million times higher. We demonstrate the combined approach in a study of proton irradiation in aluminum and employ it to predict how the depth of maximum energy deposition, the "Bragg Peak," varies depending on incident angle -- a quantity otherwise inaccessible to modelers. The lack of any experimental information requirement makes our method applicable to most materials, and its speed makes it a prime candidate for enabling quantum-to-continuum models of radiation damage. The prospect of reusing valuable TDDFT data for training the model make our approach appealing for applications in the age of materials data science.
DeepSpeed4Science Initiative: Enabling Large-Scale Scientific Discovery through Sophisticated AI System Technologies
Oct 11, 2023



In the upcoming decade, deep learning may revolutionize the natural sciences, enhancing our capacity to model and predict natural occurrences. This could herald a new era of scientific exploration, bringing significant advancements across sectors from drug development to renewable energy. To answer this call, we present DeepSpeed4Science initiative (deepspeed4science.ai) which aims to build unique capabilities through AI system technology innovations to help domain experts to unlock today's biggest science mysteries. By leveraging DeepSpeed's current technology pillars (training, inference and compression) as base technology enablers, DeepSpeed4Science will create a new set of AI system technologies tailored for accelerating scientific discoveries by addressing their unique complexity beyond the common technical approaches used for accelerating generic large language models (LLMs). In this paper, we showcase the early progress we made with DeepSpeed4Science in addressing two of the critical system challenges in structural biology research.
14 Examples of How LLMs Can Transform Materials Science and Chemistry: A Reflection on a Large Language Model Hackathon
Jun 13, 2023



Chemistry and materials science are complex. Recently, there have been great successes in addressing this complexity using data-driven or computational techniques. Yet, the necessity of input structured in very specific forms and the fact that there is an ever-growing number of tools creates usability and accessibility challenges. Coupled with the reality that much data in these disciplines is unstructured, the effectiveness of these tools is limited. Motivated by recent works that indicated that large language models (LLMs) might help address some of these issues, we organized a hackathon event on the applications of LLMs in chemistry, materials science, and beyond. This article chronicles the projects built as part of this hackathon. Participants employed LLMs for various applications, including predicting properties of molecules and materials, designing novel interfaces for tools, extracting knowledge from unstructured data, and developing new educational applications. The diverse topics and the fact that working prototypes could be generated in less than two days highlight that LLMs will profoundly impact the future of our fields. The rich collection of ideas and projects also indicates that the applications of LLMs are not limited to materials science and chemistry but offer potential benefits to a wide range of scientific disciplines.
Cloud Services Enable Efficient AI-Guided Simulation Workflows across Heterogeneous Resources
Mar 15, 2023Applications that fuse machine learning and simulation can benefit from the use of multiple computing resources, with, for example, simulation codes running on highly parallel supercomputers and AI training and inference tasks on specialized accelerators. Here, we present our experiences deploying two AI-guided simulation workflows across such heterogeneous systems. A unique aspect of our approach is our use of cloud-hosted management services to manage challenging aspects of cross-resource authentication and authorization, function-as-a-service (FaaS) function invocation, and data transfer. We show that these methods can achieve performance parity with systems that rely on direct connection between resources. We achieve parity by integrating the FaaS system and data transfer capabilities with a system that passes data by reference among managers and workers, and a user-configurable steering algorithm to hide data transfer latencies. We anticipate that this ease of use can enable routine use of heterogeneous resources in computational science.
Colmena: Scalable Machine-Learning-Based Steering of Ensemble Simulations for High Performance Computing
Oct 06, 2021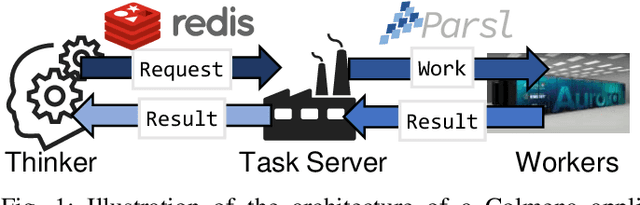
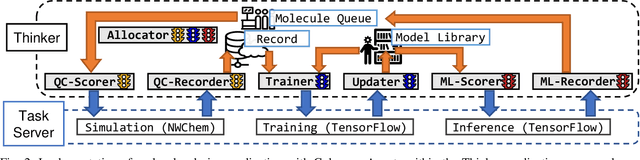
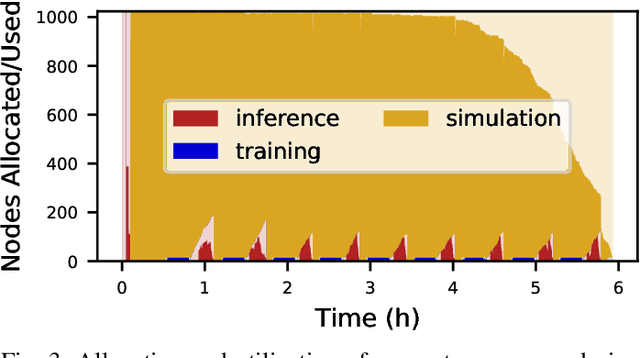
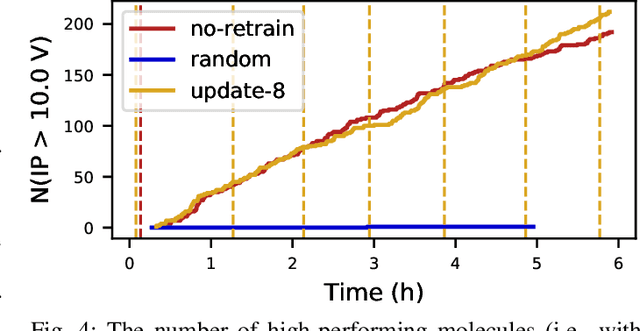
Scientific applications that involve simulation ensembles can be accelerated greatly by using experiment design methods to select the best simulations to perform. Methods that use machine learning (ML) to create proxy models of simulations show particular promise for guiding ensembles but are challenging to deploy because of the need to coordinate dynamic mixes of simulation and learning tasks. We present Colmena, an open-source Python framework that allows users to steer campaigns by providing just the implementations of individual tasks plus the logic used to choose which tasks to execute when. Colmena handles task dispatch, results collation, ML model invocation, and ML model (re)training, using Parsl to execute tasks on HPC systems. We describe the design of Colmena and illustrate its capabilities by applying it to electrolyte design, where it both scales to 65536 CPUs and accelerates the discovery rate for high-performance molecules by a factor of 100 over unguided searches.
Evening the Score: Targeting SARS-CoV-2 Protease Inhibition in Graph Generative Models for Therapeutic Candidates
May 07, 2021
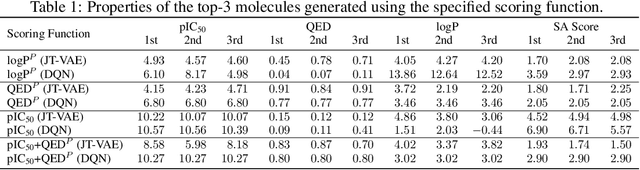

We examine a pair of graph generative models for the therapeutic design of novel drug candidates targeting SARS-CoV-2 viral proteins. Due to a sense of urgency, we chose well-validated models with unique strengths: an autoencoder that generates molecules with similar structures to a dataset of drugs with anti-SARS activity and a reinforcement learning algorithm that generates highly novel molecules. During generation, we explore optimization toward several design targets to balance druglikeness, synthetic accessability, and anti-SARS activity based on \icfifty. This generative framework\footnote{https://github.com/exalearn/covid-drug-design} will accelerate drug discovery in future pandemics through the high-throughput generation of targeted therapeutic candidates.
* arXiv admin note: substantial text overlap with arXiv:2102.04977